頭孢噻肟鈉合成工藝研究
2023-05-29 04:46:08安雪飛馮濤劉宏飛國藥集團威奇達藥業有限公司山西大同037010
化工管理 2023年15期
安雪飛,馮濤,劉宏飛(國藥集團威奇達藥業有限公司,山西 大同 037010)
1 概述
頭孢噻肟鈉(cefotaxime sodium) 是由德國Hoechst 和法國Roussel 公司聯合開發,在1977 年研制成功,于1980 年上市。頭孢噻肟鈉為白色至微黃色結晶或粉末,無臭或微有特殊臭。化學名為:(6R,7R)-3-[(乙酰氧基)甲基]-7-[2-(2-氨基噻唑-4-基)-2-(甲氧亞氨基)乙酰氨基]-8-氧代-5-硫雜-1-氮雜雙環[4.2.0]辛-2-烯-2-甲酸鈉鹽[1]。
查閱文獻和專利,頭孢噻肟鈉的合成工藝比較多。其中,原研專利GB1580623A[2]報道的合成方法是以氨噻肟酸乙酯為原料,先采用三苯基氯甲烷對氨基保護,后水解得到氨基保護的氨噻肟酸,接著在縮合劑4,5-二氰基咪唑(DCI) 存在下與7-氨基頭孢烷酸(7-ACA)縮合得到頭孢噻肟酸,最后與碳酸鈉成鹽得到頭孢噻肟鈉。該工藝采用的三苯基氯甲烷毒性較強、腐蝕性較大,縮合劑DCI 在體系中難除盡,影響產品的質量控制。目前,最常用的合成方法為7-ACA與AE 活性酯縮合得到頭孢噻肟酸,再與醋酸鈉成鹽得到頭孢噻肟鈉[3]。反應路線如圖1 所示。
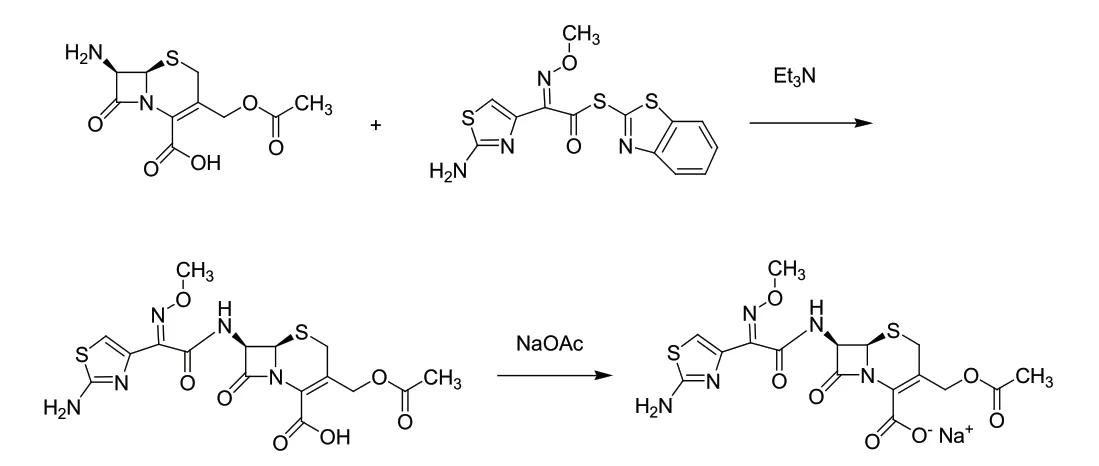
圖1 頭孢噻肟鈉反應路線圖
基因毒性雜質是指能引起基因毒性的雜質,包括致突變性雜質和其他類型的無致突變性雜質[4]。基因毒性物質可以在很低濃度時即可造成人體遺傳物質的損傷,進而導致基因突變并可能促使腫瘤發生。因其毒性較強,對用藥的安全性產生了強烈的威脅,近年來也越來越多的出現因為在已上市藥品中發現痕量的基因毒性雜質殘留而發生大范圍的醫療事故,被FDA 強行召回的案例,給藥廠造成了巨大的經濟損失[5]。各國的法規機構如ICH、FDA、EMA 等都對基因毒性雜質有了更明確的要求,越來越多的藥企在藥品研發過程中也著重關注了基因毒性雜質的控制和檢測。2-巰基苯并噻唑是一種淡黃色單斜針狀或葉片狀結晶。2017 年10 月27 日,世界衛生組織國際癌癥研究機構公布的致癌物清單初步整理參考,2-巰基苯并噻唑在2A 類致癌物清單中。以AE 活性酯為原料生產頭孢噻肟酸產品副產物2-巰基苯并噻唑,若不對頭孢噻肟酸中的2-巰基苯并噻唑進行控制,將會影響原料藥頭孢噻肟鈉中2-巰基苯并噻唑的殘留。根據 EMEA 人用藥品委員會(CHMP)關于基因毒性雜質的限度指南規定及頭孢噻肟鈉最大日劑量可以計算得出,2-巰基苯并噻唑每日允許攝入量為 5 μg/g[6]。
本文通過對反應溶劑、AE 活性酯用量,縛酸劑用量的篩選,結晶pH 值以及打漿工藝的優化確定了頭孢噻肟酸的合成工藝。該工藝生產的頭孢噻肟酸在保證質量和收率的前提下,降低了產品中2-巰基苯并噻唑的含量,進而控制頭孢噻肟鈉中2-巰基苯并噻唑的殘留。而在頭孢噻肟鈉的合成過程中通過二異丙胺的加入和結晶溫度的控制,抑制雜質的降解,提升產品的質量。
2 實驗部分
2.1 試劑與儀器
試劑:7-ACA(7-氨基頭孢烷酸),工業級,國藥集團威奇達藥業有限公司;AE 活性酯,工業級,河北合佳醫藥科技集團股份有限公司;醋酸鈉,工業級,山西繁榮富化工有限公司;四氫呋喃、二氯甲烷、三乙胺、鹽酸、丙酮、活性炭、乙酸乙酯、甲醇等未市售分析級。
儀器:pH 計(METTLER TOLEDO);電子天平;循環水式真空泵;安捷倫液相色譜儀1260;調頻電動攪拌器;自動水分測定儀(METTLER TOLEDOD)。
2.2 實驗過程
在反應瓶中投入800 mL 四氫呋喃、200 g 純化水,降溫至0~5 ℃,加入100 g 7-ACA,攪拌10 min,滴加39.4 g 三乙胺,滴完攪拌至體系逐漸變清,加入140 g AE 活性酯,控溫0~5 ℃攪拌反應至中控檢測7-ACA含 量≤0.5%,加 入1 g 活 性 炭,攪 拌10~30 min,抽濾,用20 mL 四氫呋喃淋洗,料液轉移至2 000 mL反應瓶,緩慢滴加15% 鹽酸將pH 值控制在2.5~3.0 時停止滴加鹽酸,控溫0~5 ℃析晶1 h,復測pH 值在2.0~3.5,抽濾,用200 mL 丙酮分兩次泡洗,抽干得濕品。所得濕品加入1 000 mL 丙酮打漿一次,攪拌20~30 min,抽濾,用丙酮淋洗,抽干得濕品,濕品40 ℃減壓干燥得干品頭孢噻肟酸。
在反應瓶中投入80 g 甲醇、10 g 純化水,降溫至-5~0 ℃,加入30 g 乙酸乙酯、3.6 g 二異丙胺,分批加入60 g 頭孢噻肟酸,控溫-5~0 ℃,加入10.8 g無水乙酸鈉,保溫反應30 min,繼續滴加100 g 乙酸乙酯,加晶種,保溫攪拌1~2 h;第二次滴加100 g 乙酸乙酯,約1~2 h,滴完保溫-5~0 ℃攪拌30 min;第三次滴加800 g 乙酸乙酯,約30~60 min,滴完保溫-5~0 ℃攪拌30 min;抽濾,用乙酸乙酯淋洗,抽干得濕品,濕品40 ℃減壓干燥得干品頭孢噻肟鈉。
2.3 結果與討論
2.3.1 反應溶劑的篩選
目前文獻報道的頭孢噻肟酸合成工藝[7-8],一般采用四氫呋喃-水體系、二氯甲烷-醇體系或者二氯甲烷單一溶劑。本文從收率方面,對四氫呋喃-水體系、二氯甲烷-異丙醇體系、二氯甲烷-乙醇體系和二氯甲烷體系進行了比較,對應的摩爾收率分別為97.94%、74.38%、80.75% 和65.86%。結果顯示:采用四氫呋喃/水體系的收率最高。
2.3.2 AE 活性酯用量的篩選
從合成路線可以看出,AE 活性酯是制備頭孢噻肟酸的關鍵物料,其用量既涉及工藝的成本控制,又關乎產品的質量。頭孢噻肟酸的合成一般都是AE 活性酯過量。因此,以7-ACA 為基準,考察兩個物料摩爾比對反應的影響。實驗結果如表1 所示。
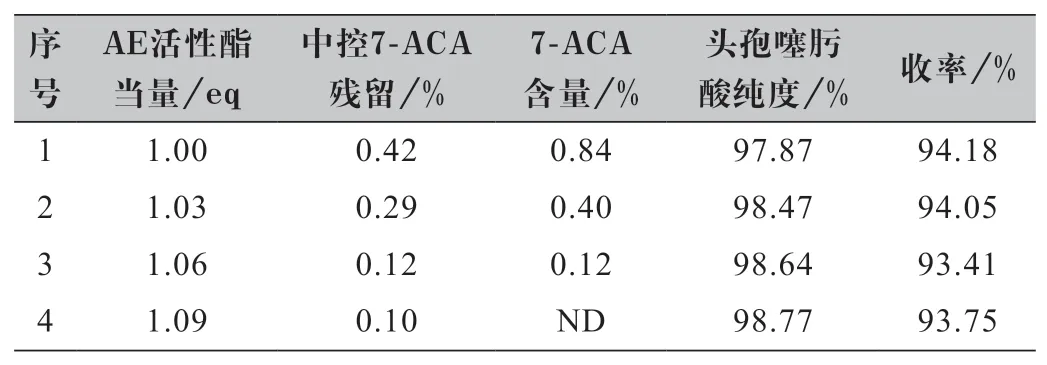
表1 AE 活性酯用量篩選實驗結果
結果顯示:隨著AE 活性酯用量的增加,7-ACA在頭孢噻肟酸中的殘留逐漸降低至零,頭孢噻肟酸純度逐漸提高,收率基本保持不變,考慮到7-ACA 的殘留在成品頭孢噻肟鈉中去除率不高,從質量角度出發,優選7-ACA:AE 活性酯的摩爾比1.00∶1.09。
2.3.3 縛酸劑用量的篩選
查閱文獻頭孢噻肟酸的合成一般采用三乙胺為縛酸劑[9],實驗中嘗試其他種類堿性化合物N,N-二異丙基乙胺、吡啶、二乙胺、5%NaHCO3、5%NaOH 等均存在7-ACA 殘留偏高的問題,因此對三乙胺的用量進行了優化,實驗結果如表2 所示。
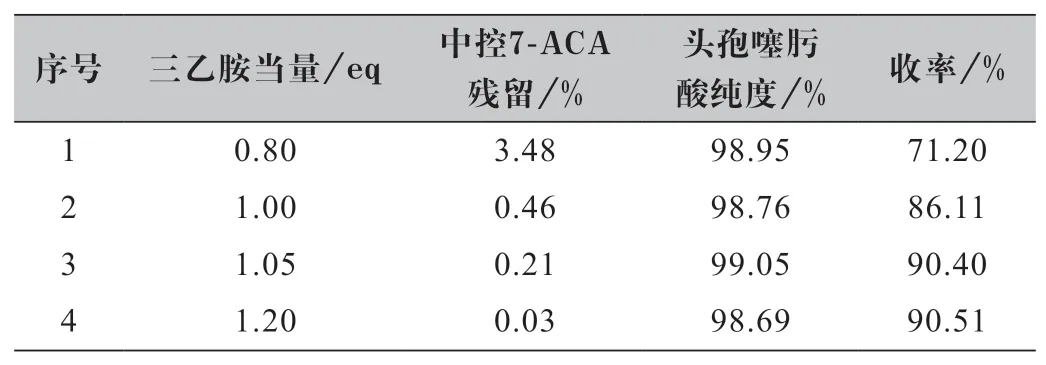
表2 三乙胺用量篩選實驗結果
實驗結論:通過表2 分析可知,當三乙胺用量≤1.00 當量時,反應原料不能夠完全溶解,原料7-ACA 轉化不夠徹底,殘留偏大,使得反應收率降低,但對產品純度影響不大;當三乙胺用量達到1.05 當量時,體系能夠溶清,反應基本進行完全,產品純度和收率均較高,繼續增加三乙胺用量收率基本保持不變。因此,從成本角度出發,優選三乙胺的用量為1.05 當量。
2.3.4 結晶pH 值得篩選
通過頭孢噻肟酸的反應機理可知,反應形成的頭孢噻肟酸在三乙胺的存在下以有機堿的鹽形式存在而溶于四氫呋喃和水溶劑中,通過滴加鹽酸進行酸化,頭孢噻肟酸以固體形式析出。從理論上分析,鹽酸只起到中和三乙胺的作用,只要三乙胺被完全中和,頭孢噻肟酸就能夠完全析出,因此鹽酸用量應與三乙胺摩爾量相當,結合對頭孢噻肟酸本身的產品pH 值約為2.6,可以推斷出結果過程體系的pH 值應該控制在2.6 左右為宜,但是基于體系組分的復雜性和頭孢噻肟酸在酸性條件下的不穩定性,酸化不同的pH值可能影響產品的質量和收率,因此對結晶pH 值進行考察。實驗結果顯示:結晶pH 值的高低是由鹽酸用量決定的,當鹽酸用量較大,pH 值低于2.0 時,產品收率越低,這主要是由于頭孢噻肟酸結構中還存在氨基官能團,會與過量的鹽酸成鹽而溶于母液中,從而導致收率降低;當鹽酸用量不足,pH 值較高處于3.0~4.0 之間,此時部分三乙胺未被中和,仍有部分頭孢噻肟酸以三乙胺鹽形式存在于母液中,因此收率仍會有所降低;當鹽酸用量于三乙胺用量相當時,此時體系pH 值在2.5~3.0 之間收率最高,考慮到實際生產過程操作的方便性,鹽酸的濃度會存在一定的波動性,以用量去調節pH 值會存在較大的差異,因此,通過監控pH 值更加容易實現,pH 值控制在2.5~3.0 之間為宜。
2.3.5 打漿工藝的優化
通過資料查閱和實驗可知[10],基因毒性雜質2-巰基苯并噻唑在母液和丙酮中有較好的溶解性,因此選擇丙酮打漿考察其去除效果。分別對頭孢噻肟酸處理前的母液、丙酮一次打漿料液和二次打漿料液中2-巰基苯并噻唑含量進行了檢查,實驗結果顯示,2-巰基苯并噻唑在處理前母液中含量為1 944 mg/L,經丙酮一次打漿后含量降為690 mg/L,二次打漿后降為200 mg/L,去除率分別為64.51% 和71.01%。同時,將頭孢噻肟酸轉成頭孢噻肟鈉后,2-巰基苯并噻唑含量同樣有降低趨勢,由處理前的28 mg/L,經丙酮一次打漿和二次打漿后降為4 mg/L 和2 mg/L。
實驗結論:通過丙酮打漿方式可以有效地降低頭孢噻肟酸中基因毒性雜質2-巰基苯并噻唑的殘留量。進一步地研究頭孢噻肟酸中2-巰基苯并噻唑殘留量對頭孢噻肟鈉中2-巰基苯并噻唑殘留量的影響,發現當頭孢噻肟酸中2-巰基苯并噻唑超出1 000 mg/L,頭孢噻肟鈉中2-巰基苯并噻唑就會超出標準5 mg/L,因此采用丙酮打漿方式一次即可滿足要求。
2.3.6 二異丙胺對頭孢噻肟鈉質量的影響
由于頭孢噻肟鈉結構中存在β-內酰胺環結構,其結構的穩定性較差,故頭孢噻肟鈉合成步驟的關鍵是控制好反應條件防止產物的降解雜質產生,而緩和劑二異丙胺的加入對抑制產物降解起到很好的作用[11],但二異丙胺的加入會導致最終產品中出現殘留。本文對二異丙胺用量進行研究,結果如表3 所示。
實驗結論:通過表3 分析可知,未使用緩和劑二異丙胺的樣品雜質明顯偏大,超出0.10% 的鑒定閾值,使用二異丙胺的批次樣品雜質明顯減小,但二異丙胺用量不宜過大,用量越多其成品中殘留越大,結合雜質和二異丙胺殘留綜合考量,二異丙胺用量為0.3 當量左右較為適宜。
2.3.7 頭孢噻肟鈉結晶溫度對產品質量的影響
由于頭孢噻肟鈉結構中存在β-內酰胺環結構,其結構的穩定性較差,故在其制備過程中的控制好溫度條件防止降解雜質的產生是關鍵因素。對頭孢噻肟鈉結晶溫度條件進行考察,結果發現:當結晶溫度超過30 ℃時,出現反應顏色加深,晶體團聚的現象,雜質也明顯增大,產品純度大幅度降低;當溫度控制在-10~30 ℃時,結晶過程晶體團聚現象消失,料液顏色隨著溫度的降低逐漸變淺,產品雜質也呈逐漸降低趨勢。同時,在實驗中還發現結晶溫度對產品晶型也有明顯影響,進而影響產品的粒度和溶劑殘留,當溫度控制在-10~10 ℃時,產品的粒度一致性最佳,產品晶型與原研參比制劑一致,溶劑殘留可得到有效地控制;當溫度降低至-10 ℃以下出現結冰現象。因此,綜合生產成本和產品質量情況考慮,結晶溫度控制在5~10 ℃最佳。對自制的產品與參比制劑進行X射線粉末衍射檢測,表明參比制劑與實驗樣品X射線粉末衍射圖譜一致,檢測結果對比如圖2 所示。
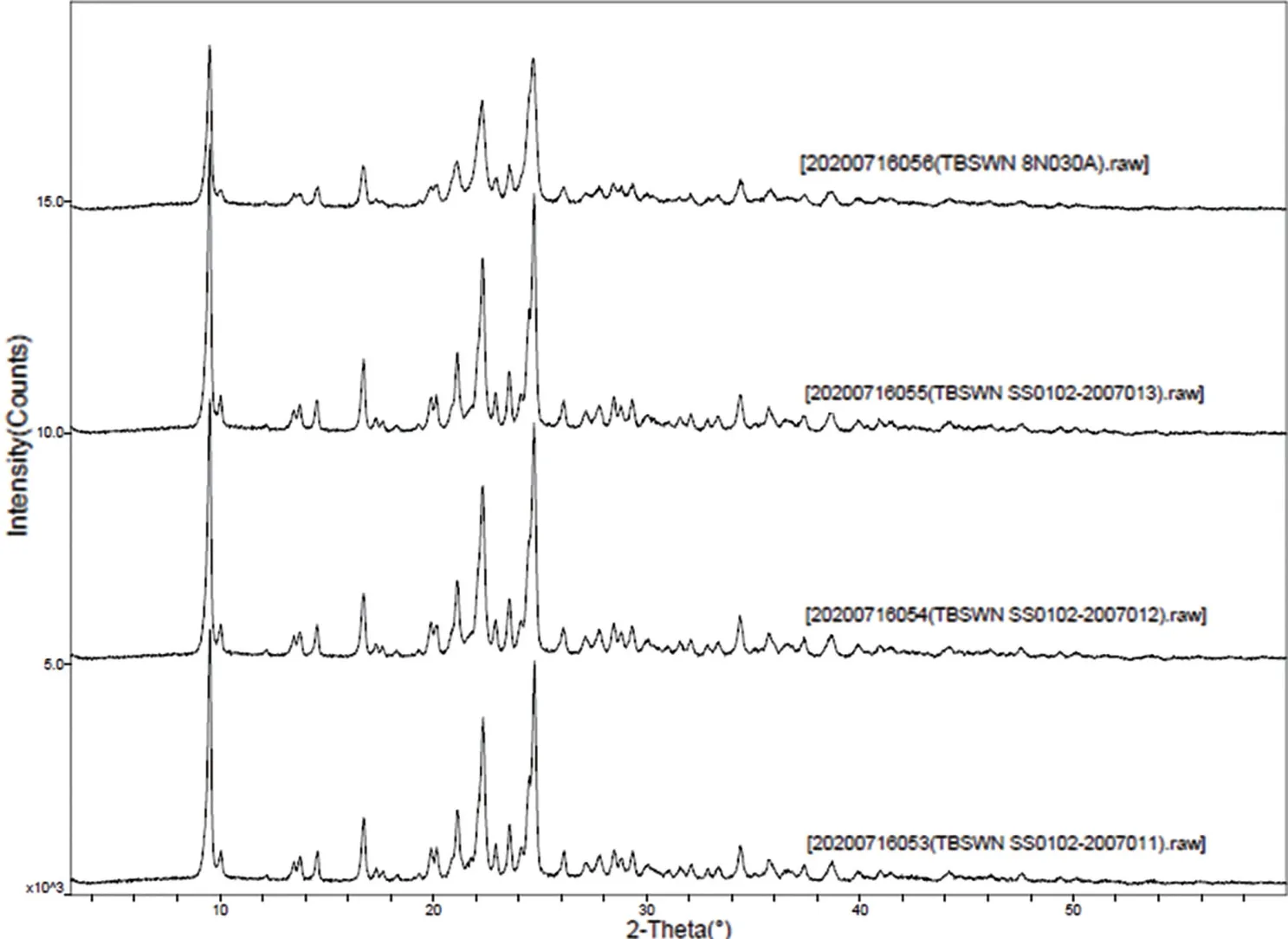
圖2 參比制劑與實驗樣品X射線粉末衍射圖譜對比
2.3.8 討論
以7-ACA 和AE 活性酯為原料,三乙胺為縛酸劑制備頭孢噻肟酸粗品,后用丙酮打漿得到頭孢噻肟酸。其中,溶劑采用四氫呋喃-水體系、7-ACA 和AE活性酯的摩爾比優選1.00∶1.09、三乙胺的用量優選1.05 當量,結晶pH 值控制在2.5~3.0 之間,配合一次丙酮打漿可得到2-巰基苯并噻唑含量低于1 000 mg/L的頭孢噻肟酸。
在頭孢噻肟鈉的合成過程中通過加入二異丙胺,可以明顯改善料液的穩定性,將結晶溫度控制在5~10 ℃,對提升產品質量,控制產品的晶型和粒度,改善產品溶劑殘留等有明顯的作用,且產品中的基因毒性雜質2-巰基苯并噻唑含量可以穩定地控制在限度之內。該工藝生產周期短,反應條件溫和、收率高、產品質量好、適宜規模化工業生產。
