頭頸部Castleman病四例臨床分析*
2023-05-08 00:32:52朱登耀何曉光蔣東輝楊聲豪賈延軒
云南醫藥 2023年2期
關鍵詞:手術
朱登耀,何曉光,蔣東輝,楊聲豪,賈延軒,徐 昕
(昆明醫科大學第一附屬醫院 頭頸外科,云南 昆明 650032)
1 臨床資料
1.1 一般情況
4例患者,其中三例男性,1例女性。病變部位多見于頸部左側,平均年齡及中位年齡均為29歲(24~39歲),初次就診到確診平均為1.25個月(0.5~2月)
1.2 臨床表現
所有患者均以無誘因出現頸部無痛性淋巴結腫大為首發癥狀,頸部觸診可觸及邊界清楚、活動欠佳的腫物,伴觸壓痛,其中一例伴有呼吸困難,其余三例無呼吸困難、咳嗽、發熱等表現。
1.3 輔助檢查
所有患者入院行胸片、心電圖、生化檢查均無特殊異常,四例頸部增強CT分別提示:左側胸鎖乳突肌內測至胸廓入口,邊界清楚,大小約4.3*9.2*3.5 cm(見圖2中的A、B、D圖);左側II區類圓形結節狀軟組織密度影,大小約2.5*3.5*2.6 cm;左側頸動脈鞘后方見邊界清楚的軟組織腫塊影,大小約3.1*8.3*5.2 cm;左頸部淋巴結腫大,大小約2.8*3.6*3.1 cm,邊界清楚。
1.4 診治方法
排除手術及麻醉禁忌癥后,所有患者在全麻下行“頸部軟組織病損切除術+功能性頸部淋巴結清掃術”。術后行抗炎、補液等治療。術后隨訪2~48個月,無其他并發癥,仍在隨訪中。
1.5 術后病理檢查
術后病檢均顯示Castleman病,不同病例免疫組化結果分別出現:KI-67(+)、CD3(+)、CD20(+)、CD21(+)、CD38(+)、CD68(+)、CD138(+)、BCL-2(+)。其中一例病理分型為透明血管型(見圖1)。
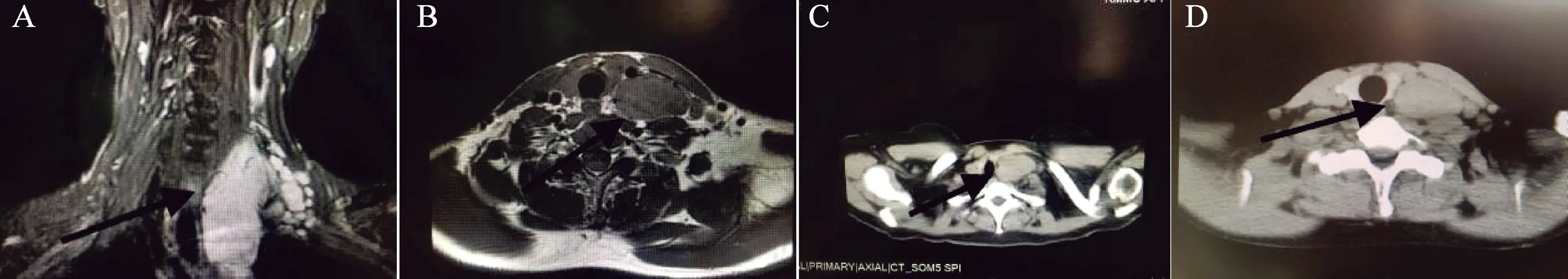
頸部CT及MRI檢查提示:頸部占位性病變明顯,邊境明顯,信號清楚,軟組織密度影。圖2 典型頸部Castleman病患者影像學資料
2 討論
本文主要討論頭頸部Castleman病的臨床特性以及治療方式,以提高頭頸部Castlema病的治療水平。Castleman病是一種罕見的淋巴組織增生性疾病,于1956年由Castleman發現并命名[1],該病可發病于任何年齡段,多見于10~45歲,男女無顯著差異,發病部位以縱隔、腹部為主,較少發生于頭頸部[2]。臨床上根據其受累淋巴結分為中心型和多中心型。根據其病理特征分為透明血管型、漿細胞型、混合型。其中中心型病理表現多為透明血管型。該病目前病因尚未明確。無論是中心型還是多中心型發病機制主要是由白細胞介素-6信號通路增加引起的,且白細胞介素-6受體多態性在HIV陰性的Castleman病中普遍存在,并與可溶性白細胞介素-6受體水平升高相關[3]。所以如何控制白細胞介素-6指標的含量為臨床治療方案提供了一種新的思路。中心型的臨床表現無特異性,是一個典型生長緩慢腫塊,雖然增大的淋巴結可能壓迫重要組織器官,但中心型很少危及生命,以本文四組患者為例,四例患者均是無意間發現頸部腫塊,本組四例患者在術前,完善實驗室生化檢測、影像學檢查后,僅有一例懷疑為本病,其余患者皆未做出明確診斷。因在頸部巨大腫物中,頸部神經鞘瘤往往也是體積較大、臨床癥狀相似,多常起源于頸動脈間隙,所以術前重點判斷該頸部腫物與神經鞘瘤的區別,中心型影像學CT增強特征;(1)早期和持續強化;(2)強化均勻與否與腫塊大小相關,腫塊越大則強化越均勻。且少數神經鞘瘤臨床表現中伴有疼痛感,而神經鞘瘤影像學CT增強密度低于肌肉、常有包膜、腫瘤形態較大時易發生囊變。所以當遇到頸深部不明原因的巨大腫物,醫師應重點鑒別這兩者之間的影像學特點。中心型與多中心型在治療上是截然不同的,中心型病變位置局限,臨床特征除明顯包塊以外無其他有價值表現,通過手術切除通常治愈且預后良好,本文四位患者術后隨訪至今,無一例復發轉移,因此手術是中心型首選的一線治療,有兩點是需要在術前考慮的:一是淋巴結腫大的位置。二是腫大淋巴結的可切除性,以及腫大淋巴結可能通過壓迫鄰近解剖結構或誘發全身炎癥綜合征而引起癥狀的程度,還應進一步考慮目前有無癥狀,但如果腫塊繼續增長,可能通過壓迫鄰近結構而成為癥狀的可能性。可切除性是一個主觀的決定,術者應該權衡手術的風險,并由外科醫生和患者協商決定,鼓勵多學科討論,以降低患者手術風險和最大程度改善患者預后。因壓迫鄰近結構或腫塊體積過大以及無法耐受手術的患者,可通過藥物治療,如利妥昔單抗、類固醇等、放療或栓塞進行切除[4-6]。多中心型由于全身表現眾多,病情復雜,目前尚無統一的治療方案,多以化療藥物為主手術為輔且療效及預后有比較大的差異,臨床上多采用糖皮質激素或聯合化療(CHOP方案)[7]。頭頸部Castleman病作為罕見病,應該作為頸部腫物鑒別的對象,該病相關分型的發病機制還有待深入研究,相信隨著研究機制的深入,會加深一線臨床醫生對Castleman病的了解,治療方案也將提高,患者預后也會得進一步的改善。
猜你喜歡
環球時報(2022-12-23)2022-12-23 09:28:37
昆明醫科大學學報(2022年1期)2022-02-28 07:45:04
中老年保健(2021年11期)2021-08-22 03:13:36
昆明醫科大學學報(2021年2期)2021-03-29 07:42:46
河北畫報(2020年10期)2020-11-26 07:20:50
小學閱讀指南·低年級版(2017年1期)2017-03-13 20:07:35
中國衛生標準管理(2015年3期)2016-01-14 03:41:47
中國醫療美容(2015年1期)2015-07-12 10:06:38
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:54
西南軍醫(2014年5期)2014-04-25 07:42:48
