濁點萃取—異辛烷反萃取—氣相色譜質譜法測定茶飲料中8種農藥殘留
2023-03-22 13:02:34孔令燦孟元華
食品與機械 2023年1期
關鍵詞:方法
謝 潔 周 閏 孔令燦 孟元華 王 宇
(1.無錫市疾病預防控制中心,江蘇 無錫 214023;2.南京醫科大學,江蘇 南京 211166)
隨著生活節奏的不斷加快,人們對茶產品的要求逐漸向方便化趨勢發展,茶,正以時尚化、便捷化的方式進入人們的生活。隨著茶飲料的大量普及,相應的安全問題也受到社會的大量關注,如農殘檢測[1]。在茶飲料中,農殘含量相對較低,按照目前傳統的檢測方法,在檢測中需要用到大量的有機溶劑,更需要花費大量時間在濃縮提取過程上,操作繁瑣,費時費力。
國內外對于農藥殘留的分析報道中,儀器方法主要有:免疫分析技術[2]、氣相色譜[3]、毛細管電泳技術[4]、氣相色譜—質譜聯用[5-6]、液相色譜—質譜聯用[7]、超高效液相色譜—質譜聯用[8-9]等。其中,樣品前處理技術多為分散固相萃取[10-11]、分散液液微萃取[12]、固相萃取柱凈化[6]、QuEChERS凈化[13-14]等,在操作過程中均需使用大量有機溶劑,耗時長,操作步驟繁瑣,且需要復雜的凈化步驟。濁點萃取法(cloud point extraction,CPE)能在一定程度上避免上述問題,研究擬以表面活性劑水溶液作為萃取劑,通過表面活性劑的增溶性和濁點分相來對目標物進行分離和富集,表面活性劑中的目標物則通過微量的有機溶劑萃取出來,該方法不但能提高富集倍數,同步完成待測組分的提取和凈化,也能有效避免表面活性劑的高黏度對檢測儀器產生的影響。該技術目前已被廣泛應用于重金屬分析[15]、食品工業[16-17]、金屬離子形態分析[18-19]、環境污染物檢測[20-21]、環境有機分析[22]等領域,但是關于濁點萃取反萃取法同時富集分離茶飲料中多種農藥的研究尚未見報道。
研究擬以非離子表面活性劑PEG 4000作為萃取劑從茶葉飲料中提取和預濃縮8種農殘,再從獲得的富含表面活性劑相中反萃取到異辛烷中。從而建立從茶飲料中同時提取檢測多種農藥殘留的濁點萃取方法,以期為茶飲料中擬除蟲菊酯和有機氯類農藥殘留的檢測提供參考。
1 材料與方法
1.1 材料
1.1.1 主要儀器
氣相色譜/質譜儀:7890B-7000C型,美國Agilent公司;
超純水系統:Milli-Q型,美國Millipore公司;
高速冷凍離心機:3-30K型,美國Sigma公司;
電子天平:XS105DU型,美國Mettler Toledo公司;
超聲儀:SCQ-10018型,上海聲彥超聲波儀器公司;
渦旋混合器:G560型,美國Scientific Industries公司。
1.1.2 試劑與材料
艾氏劑(CAS:309-00-2)、狄氏劑(CAS:60-57-1)、六氯苯(CAS:118-74-1)、七氯(CAS:76-44-8)、氯氰菊酯(CAS:52315-07-8)、甲氰菊酯(CAS:64257-84-7)、溴氰菊酯(CAS:52918-63-5)、氰戊菊酯(CAS:51630-58-1):純度≥99.5%,德國Dr.Ehrenstorfer公司;
正己烷:色譜純,德國默克公司;
異辛烷:色譜純,上海阿拉丁生化科技股份有限公司;
石油醚:色譜純,美國TEDIA公司;
茶飲料6種:市售;
PEG 4000:分析純,韓國樂天公司;
Tween-20:分析純,德國默克公司;
Triton X-100:分析純,美國Sigma公司;
氯化鈉、無水硫酸鈉:分析純,國藥集團化學試劑有限公司;
試驗用水:符合GB/T 6682—2008《分析實驗室用水規格和試驗方法》規定的一級水。
1.2 方法
1.2.1 農藥標準溶液的配制 分別取適量8種農藥標準品,用正己烷配制成質量濃度為5 mg/L的4種有機氯類農藥和質量濃度為10 mg/L的4種擬除蟲菊酯類農藥的混合標準中間液,-20 ℃保存備用,有效期為6個月。再用異辛烷逐級稀釋,有機氯類農藥逐級稀釋為2.0,1.0,0.5,0.1,0.05,0.01 mg/L的混合標準儲備液,擬除蟲菊酯類農藥逐級稀釋為4.0,2.0,1.0,0.2,0.10,0.02 mg/L的混合標準儲備液,均保存于4 ℃,有效期為2~3周。
1.2.2 樣品前處理
(1)濁點萃取方法:采集市場上不同品牌的6個茶飲料樣品,準確移取10.0 mL于25 mL刻度試管中,依次準確加入300 g/L的PEG 4000溶液2.0 mL、無水硫酸鈉1.0 g,混合均勻后經超聲輔助至全部溶解。將上述混合溶液于40 ℃水浴20 min。待其靜置分層后用長細針頭的注射器吸出水相棄去。
(2)反萃取方法:向剩下的表面活性劑相中準確加入異辛烷200 μL,漩渦混合1 min,待兩項分層后,移液槍吸取上層異辛烷相至自動進樣小瓶的內襯管中,待測。
(3)加標回收方法:取空白茶飲料18份,分別添加低、中、高3種不同濃度(六氯苯、七氯、艾氏劑、狄氏劑4種農藥的低、中、高質量濃度為0.01,0.10,1.0 μg/L,聯苯菊酯、甲氰菊酯、高效氯氟氰菊酯、氯氰菊酯4種農藥的低、中、高質量濃度為0.02,0.20,2.0 μg/L)的標準溶液進行加標回收試驗,每個加標水平重復6次。
1.2.3 色譜條件 進樣口溫度280 ℃;進樣量1 μL,不分流進樣;恒流模式,流速1 mL/min;Agilent HP-5 ms UI毛細管色譜柱(30 m×0.25 mm×0.25 μm);柱升溫程序:60 ℃保持2 min,以20 ℃/min升溫至120 ℃并保持1 min,以10 ℃/min升溫至250 ℃并保持3 min。
1.2.4 質譜條件 在選擇性離子(SIM)模式下檢測,采用電子轟擊源(EI+);電壓70 eV;傳輸線溫度150 ℃,離子源溫度230 ℃,四極桿溫度150 ℃,溶劑延遲時間10 min。
1.2.5 數據處理 采用安捷倫Mass Hunter workstation軟件進行提取保留時間和色譜峰面積,每組試驗重復測定的結果以回收率和相對標準偏差,用Microsoft Office Excel 2017軟件進行數據處理。使用Origin 8.0繪制譜圖。
2 結果與分析
2.1 方法性能指標
配制8種農藥不同濃度的系列混合標準溶液,經質譜分析,8種農藥的保留時間、定性離子、定量離子(見表1)色譜峰均獲得良好分離,峰形良好。標準色譜圖(有機氯類農藥質量濃度為1.0 mg/L的標準溶液,擬除蟲菊酯類農藥質量濃度為2.0 mg/L的標準溶液)如圖1所示。
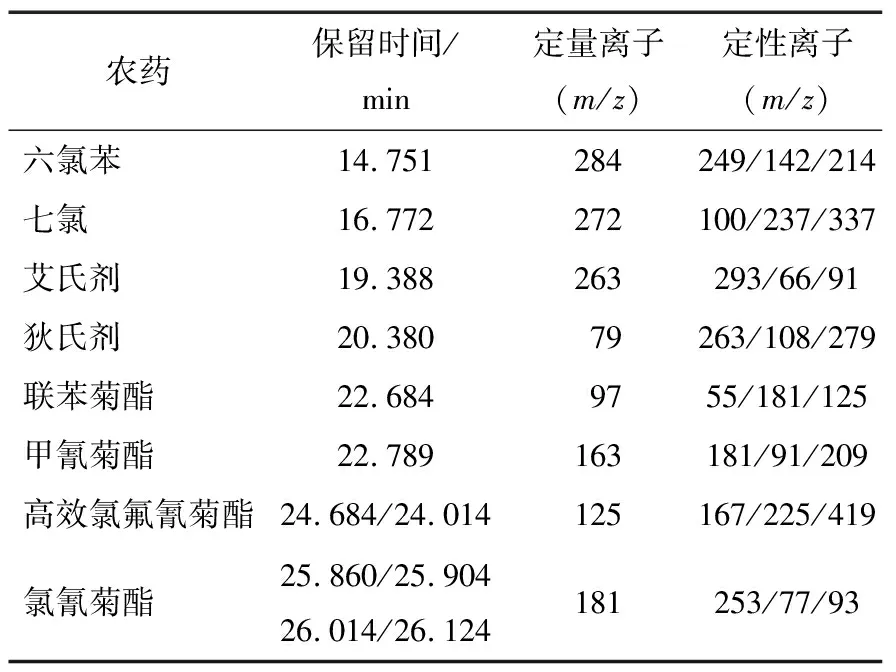
表1 8種農藥保留時間和SIM掃描離子

1.六氯苯 2.七氯 3.艾氏劑 4.狄氏劑 5.聯苯菊酯 6.甲氰菊酯 7.高效氯氟氰菊酯 8.氯氰菊酯 有機氯類農藥質量濃度為1.0 mg/L 擬除蟲菊酯類農藥質量濃度為2.0 mg/L
2.2 濁點萃取優化
2.2.1 表面活性劑種類以及用量的選擇 選擇Tween-20、Triton X-100和PEG 4000 3種表面活性劑作為萃取劑時,8種農藥的回收率。結果發現,Tween-20和Triton-100為萃取劑時,未能與反萃取溶劑異辛烷分層或分層不明顯,無法取上層清液進樣分析;PEG 4000為萃取劑時可與反萃取劑經離心有較好的分層,萃取效果好,故選用PEG 4000為萃取劑。考察不同用量的PEG 4000對回收率的影響,添加量分別設置為0.5,1.0,2.0,3.0 mL,回收率結果見圖2。當PEG 4000(300 g/L)添加量為0.5~2.0 mL時,8種農藥的回收率均呈上升狀態,添加量為2.0~3.0 mL時,8種農藥的回收率保持恒定,不再上升。因此,采用添加2 mL 300 g/L的PEG 4000進行萃取。
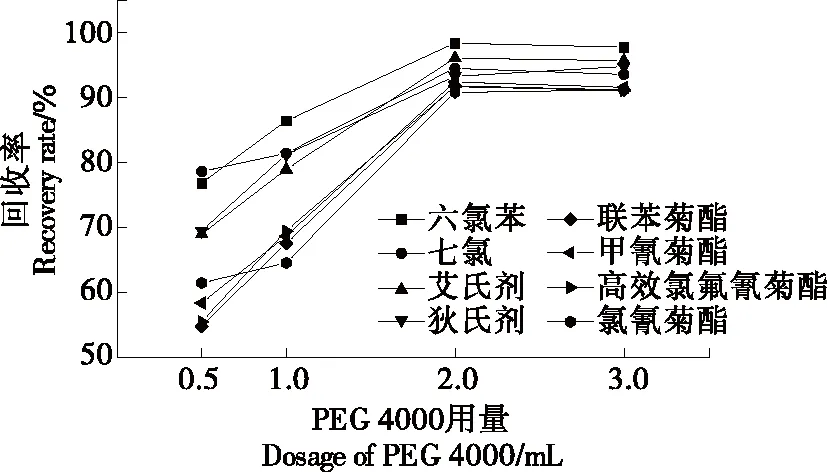
圖2 表面活性用量的選擇
2.2.2 鹽種類及用量的選擇 試驗以無水Na2SO4為惰性鹽,考察了無水Na2SO4添加量(0.5~1.2 g)對濁點萃取的影響。由圖3可知:隨著無水Na2SO4添加量的增加,萃取回收率增大,當無水硫酸鈉添加量達到1.0 g時回收率最好,此時溶液接近飽和,再增加無水硫酸鈉對回收率無影響。故試驗中所用無水硫酸鈉用量為1.0 g。
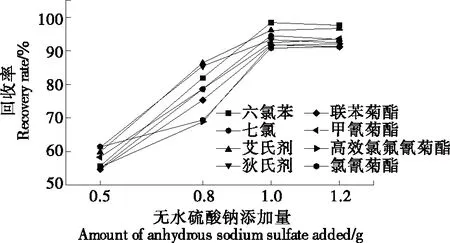
圖3 無水硫酸鈉添加量對8種農藥回收率的影響
2.2.3 萃取溫度及水浴平衡時間的選擇 萃取過程中,較高的溫度和較長的平衡時間并不會提高回收率,相反,較高的溫度和較長的平衡時間會使待測物揮發、水解,從而造成損失[20]。樣品經上述優化處理后,分別在20,30,40,50 ℃水浴平衡20 min。由圖4可知,萃取溫度為20~40 ℃時,8種農藥的回收率均呈上升狀態;萃取溫度高于40 ℃后,8種農藥的回收率均呈下降趨勢。故試驗采用的濁點萃取溫度為40 ℃。同時研究在40 ℃水浴下不同水浴平衡時間(10,15,20,30,40 min)對回收率的影響。由圖5可知,在40 ℃水浴中平衡20 min 時8種農藥的回收率最好,較高的溫度和較長的平衡時間并未提高回收率。
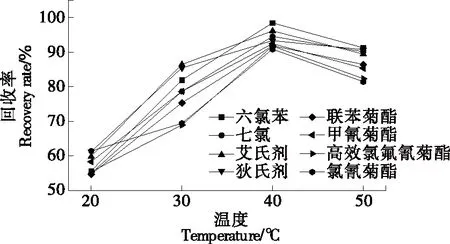
圖4 溫度對8種農藥萃取回收率影響
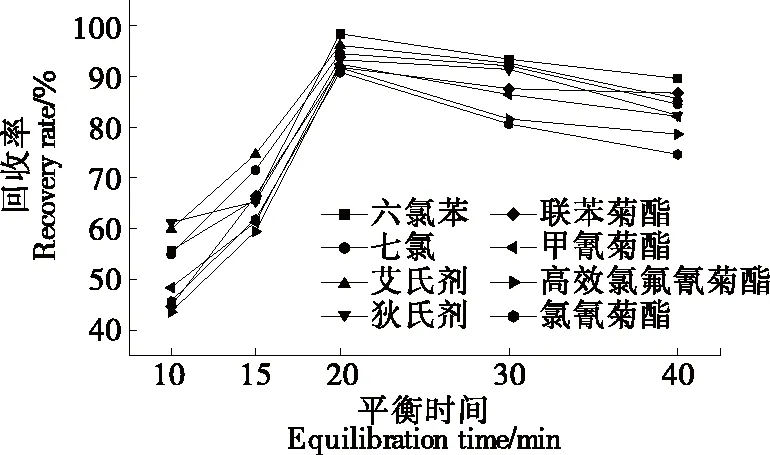
圖5 水浴平衡時間的選擇
2.3 反萃取條件優化
表面活性劑相因其性質黏稠直接進入氣質儀會造成污染,試驗考慮選擇正己烷、石油醚、異辛烷作為反萃取溶劑,由于正己烷和石油醚本身的揮發性較強,易揮發于超聲反萃取過程中,因此重現性較差,而異辛烷與表面活性劑不互溶,提取效果好,故選擇異辛烷作為反萃取劑。按上述前處理方法操作,研究異辛烷用量(100,150,200,300,500 μL)對回收率的影響,結果(見圖6)表明,采用200 μL異辛烷反萃取時,回收率較高。
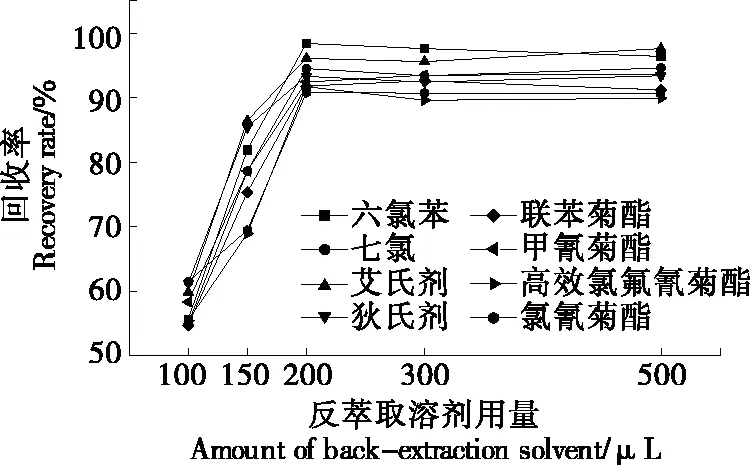
圖6 反萃取溶劑用量的選擇
2.4 方法學考察
2.4.1 線性范圍、相關系數和方法檢出限、定量限 用異辛烷將混合標準儲備液逐級稀釋成混合標準系列按上述優化好的CPE-GC/MS法,檢測得到線性范圍、線性方程、相關系數,見表2。

表2 CPE-GC/MS方法8種農藥的工作曲線、線性相關系數、檢出限和定量限
試驗采用在空白樣品中添加標準溶液按前處理后進行檢測,以最低濃度的加標回收樣液計算信噪比(S/N),以S/N=3得出檢出限(LOD),S/N=10得出定量限(LOQ)。經計算,確定該方法的檢出限為0.003~0.008 mg/L,定量限為0.01~0.02 mg/L。
2.4.2 加樣回收率和精密度試驗 吸取10.0 mL空白茶飲料18份,按照試驗優化的前處理方法和色譜條件下,每個加標濃度檢測6份,分別考察了低、中、高加標濃度下8種農藥的回收率,結果(見表3)表明,8種農藥的平均回收率為78.3%~98.4%,相對標準偏差(RSD)2.7%~8.5%,該方法的精密度和準確度均能達到滿意的結果,符合農殘檢測方法確認的要求。
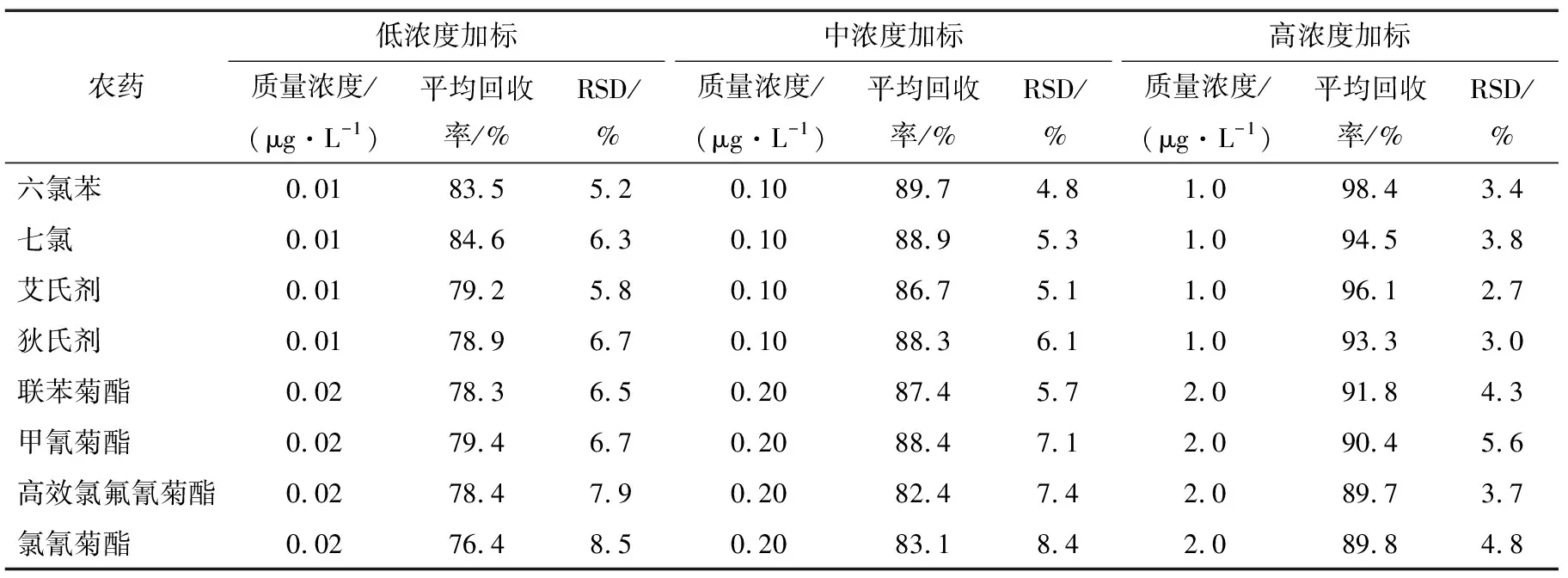
表3 3種不同加標濃度下 8種農藥的回收率和相對標準偏差
2.5 與已知方法的比較
將所建立的方法與文獻[3,5,9,11,20]報道的傳統方法從檢出限、定量限、精密度、回收率等方面進行比較,分散固相萃取、分散液液微萃取、固相萃取、QuEChERS凈化等這些傳統的前處理方法會使用到大量的有機溶劑,會對試驗人員造成損害,對環境造成污染。與其他方法相比,研究以非離子表面活性劑作為萃取劑快速有效地提取和預濃縮分離目標物,再用微量的異辛烷進行反萃取,該方法具有操作簡單,高效,富集率高,且有機溶劑用量少,整體優化了常規對茶飲料類液體飲料中農殘的提取方法。傳統農藥殘留檢測及預處理方法比較見表4。
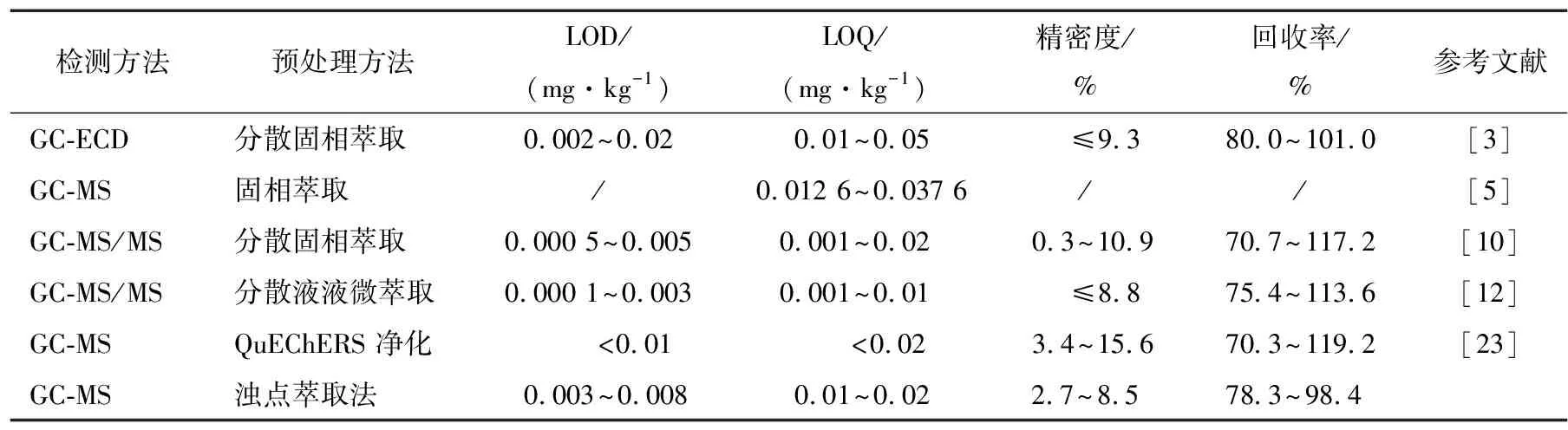
表4 與已知的農藥殘留測定方法的比較
2.6 實際樣品的檢測
采集市場上不同品牌的6個茶飲料樣品按照建立的分析方法進行農藥殘留的測定,結果均為未檢出,符合食品安全要求。
3 結論
研究通過優化表面活性劑種類及用量、鹽種類及用量、萃取溫度及平衡時間、反萃取溶劑種類及用量等前處理條件,建立了濁點萃取—異辛烷反萃取—氣相色譜質譜法測定茶飲料中8種廣泛使用的擬除蟲菊酯和有機氯類農藥(艾氏劑、六氯苯、氯氰菊酯、甲氰菊酯、溴氰菊酯等)農殘的技術。該方法分離效果好,具有良好的線性關系,相關系數R均大于0.995,方法的檢出限為0.003~0.008 mg/kg,定量限為0.01~0.02 mg/kg,平均回收率78.3%~98.4%。與傳統分散固相萃取、QuEChERS 凈化等前處理方法相比,具有快速、簡便、環保、高效、準確,有機溶劑用量少等特點,萃取過程中以表面活性劑水溶液代替有機試劑萃取目標分離物,微量異辛烷作為反萃取溶劑,降低試驗中大量有機溶劑的加入對環境的污染及試驗人員身體的損害,適用于茶飲料類液體飲品多種農藥殘留的測定。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
