水產品中5種環境激素農藥LC-MS/MS快速檢測方法
2023-03-03 13:13:16關文碧楊曉靜鄭嵋丹朱國榕
食品工業 2023年2期
關文碧 ,楊曉靜,鄭嵋丹,朱國榕
1. 肇慶學院食品與制藥工程學院(肇慶 526061);2. 農業農村部農產品質量安全風險評估實驗室(肇慶)(肇慶 526061)
在我國環境激素類農藥優先名錄中[1],三唑醇是除活體試驗以外的試驗數據證明具有內分泌干擾作用的農藥,吡蟲啉、腈菌唑、丙環唑和戊唑醇是美國環保局經4個暴露途徑篩選出的農藥品種。2018年珠江流域監測顯示吡蟲啉的檢出率為98.6%,最大檢出質量濃度為162 ng/L[2]。2019年江蘇太湖水樣監測顯示[3],戊唑醇檢出率為97%,最高檢出質量濃度為2 040 ng/L;三唑醇檢出率為100%,質量濃度為11~15 ng/L。2020~2021年間河北白洋淀中丙環唑的監測質量濃度為3.84~1 209 ng/L[4]。沈陽污水處理廠的進水和出水中均檢出腈菌唑,質量濃度在1.5~5 μg/L范圍內[5]。為保護人類健康,美國規定魚、軟體動物中吡蟲啉的限量值為50 μg/kg[6],日本規定上述5種農藥在水產品中執行0.01 mg/kg的限量標準[7]。而我國尚未對水產品中腈菌唑、丙環唑、三唑醇、戊唑醇和吡蟲啉制定最大殘留限量要求。
試驗選擇高選擇性、高靈敏度的液相色譜-串聯質譜(liquid chromatography-tandem mass spectrometry,LC-MS/MS)技術,基于高效、價廉的分散固相萃取法,建立LC-MS/MS快速檢測水產品中5種環境激素類農藥的殘留分析方法,并在廣東省內采集市售60份魚、蝦和螺樣品進行檢測,為水產品中環境激素類農藥殘留風險評估和風險預警提供數據支持。
1 材料與方法
1.1 材料與儀器
腈菌唑標準溶液100 mg/L、丙環唑標準溶液100 mg/L、三唑醇標準溶液100 mg/L、吡蟲啉標準溶液1 000 mg/L(阿爾塔科技有限公司);戊唑醇標準溶液100 mg/L(北京海岸鴻蒙標準物質技術公司);甲醇(色譜純)、乙腈(色譜純)、乙酸乙酯(分析純)、甲酸(分析純)、乙酸(分析純):上海安譜實驗科技股份有限公司;氯化鈉、無水硫酸鈉(均為分析純,西隴科學股份有限公司);PSA固相萃取填料、C18固相萃取填料(直徑40~60 μm,天津博納艾杰爾科技有限公司);γ相納米氧化鋁(直徑10 nm,南京先豐納米材料科技有限公司);弗羅里硅土(0.075 0~0.150 mm孔徑,即100~200目)、中性氧化鋁(0.048 0~0.075 0 mm孔徑,即200~300目):上海阿拉丁生化科技有限公司。
Waters Xevo TQD液相色譜質譜聯用儀(美國Waters公司);Centrisartg-16C高速冷凍離心機、Secura?分析天平(德國Sartorius公司);Multi Reax全能型振蕩器(德國Heidolph海道夫);Thermo μV-TOC超純水系統(美國Thermo公司);KQ-25ODE數控超聲波清洗器(江蘇昆山市超聲儀器公司)。
1.2 試驗方法
1.2.1 溶液配制
分別量取1.0 mL腈菌唑/丙環唑/戊唑醇/三唑醇標準溶液(100 mg/L)和100 μL吡蟲啉標準溶液(1 000 mg/L)于10 mL容量瓶中,用甲醇定容至刻度,配得10 mg/L 5種農藥的混合標準溶液。量取1 mL甲酸和1 mL乙酸分別加入100 mL容量瓶中,用乙腈定容至刻度線,搖勻,配得1.0%甲酸乙腈和1.0%乙酸乙腈。
1.2.2 樣品前處理
分別取蝦、魚、貝可食用部分,用高速攪拌機充分攪碎后置于-18 ℃冰箱中避光保存,使用前常溫解凍。稱取2 g魚、蝦及貝類樣品于50 mL塑料離心管中,加入10 mL乙腈,振蕩提取20 min,加入2 g氯化鈉和2 g無水硫酸鈉,渦旋30 s,在4 000 r/min轉速下離心5 min,取上清液過0.22 μm有機濾膜,待測。
1.2.3 色譜條件
1.2.3.1 HPLC條件
色譜柱ACQUITU UPLC? HSS T3(2.1 mm×50 mm,1.8 μm);柱溫25 ℃;流速0.3 mL/min;進樣量5.0 μL;流動相為0.1%甲酸水溶液和乙腈;洗脫方式采用梯度洗脫。洗脫程序:0~1.0 min,乙腈由10%上升至90%,1.0~3.0 min,乙腈保持90%,3.0~3.5 min,乙腈下降至10%,3.5~5 min乙腈保持10%。
1.2.3.2 質譜條件
離子化模式ESI+;質譜掃描方式采用多反應監測;干燥氣流量600 ℃;干燥溫度200 ℃;離子傳輸管溫度500 ℃;5種農藥的質譜參數見表1。
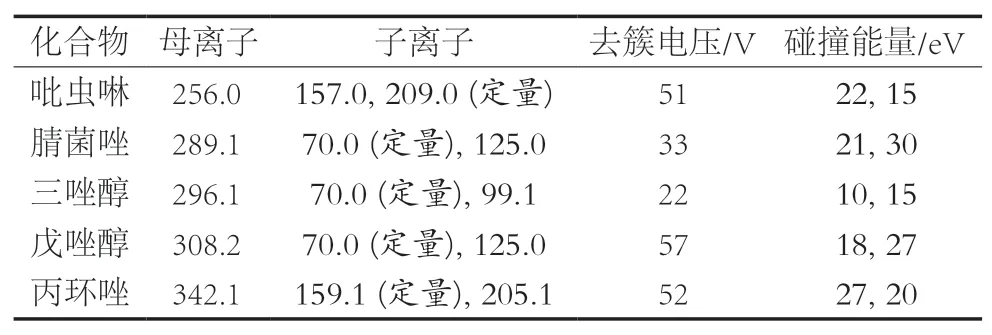
表1 5種農藥的質譜參數
2 結果與分析
2.1 樣品前處理方法的優化
2.1.1 提取溶劑的優化
試驗以蝦作為基質,選擇甲醇、乙腈、乙酸乙酯、1%甲酸乙腈和1%乙酸乙腈5種溶劑做加標回收試驗,對提取溶劑的種類進行優化,結果如圖1所示。用乙腈提取5種農藥添加回收率相對較高,目標物回收率在75%~95%,因此使用乙腈作為提取劑。
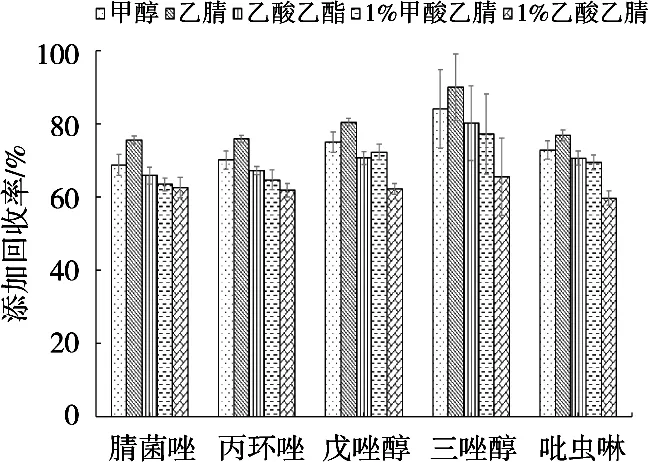
圖1 提取溶劑對蝦中農藥回收率的影響
2.1.2 提取方法優化
不同提取方式對樣品的提取效果具有一定差異,選取蝦作為樣品基質,比較振蕩提取20 min和超聲提取20 min這2種提取方式下目標物的添加回收率,結果見圖2。振蕩提取的目標物回收率比超聲提取目標物回收率高,因此,試驗選擇振蕩提取方式。

圖2 提取方式對蝦中農藥回收率的影響
2.1.3 鹽析條件的優化
試驗對比“2 g氯化鈉+2 g無水硫酸鈉”和“2 g氯化鈉+2 g無水硫酸鎂”的鹽析效果。使用“2 g氯化鈉+2 g無水硫酸鈉”作為鹽析劑,5種農藥回收率為75.7%~91.8%,SRSD為3.38%~7.59%。使用“2 g氯化鈉+2 g無水硫酸鎂”作為鹽析劑,5種農藥回收率為69.3%~114.2%,CV為4.11%~11.21%。所以試驗選用“2 g氯化鈉+2 g無水硫酸鈉”作為鹽析劑。
2.1.4 凈化條件的優化
試驗分別以50 mg PSA、50 mg C18、50 mg 10 nm氧化鋁、50 mg弗羅里硅土和50 mg中性氧化鋁作為凈化劑,以及不加凈化劑6個條件進行凈化效果的對比。不同凈化條件下蝦中農藥添加回收試驗結果如圖3所示。使用等量的PSA、C18、納米氧化鋁、弗羅里硅土和中性氧化鋁時,吡蟲啉添加回收率分別為67.1%,61.3%,66.6%,59.4%和66.7%。不使用凈化劑時,吡蟲啉回收率為78.2%。凈化劑的使用會導致吡蟲啉的添加回收率下降。同時,使用C18作為凈化劑會使丙環唑和戊唑醇下降到66.4%和69.6%。此外,對比不同凈化條件下目標物的基質效應。基于1.2.2的樣品前處理方法,分別以50 mg PSA、50 mg 10 nm氧化鋁、50 mg弗羅里硅土和50 mg中性氧化鋁作為凈化劑對1 mL空白蝦樣品提取溶液進行凈化及不加凈化劑凈化,得到不同凈化條件下蝦的空白樣品溶液,用以稀釋10 mg/L的5種農藥的混合標準溶液,配制5.0~200 μg/L的基質標準溶液和溶劑標準溶液。按照式(1)計算農藥的基質效應,結果如表2所示。無凈化和加入不同凈化劑對目標農藥的基質效應影響不大。因此,在滿足回收率的要求并兼顧節省經濟成本、簡化提取步驟的前提下,試驗選擇省略添加凈化劑步驟,樣品經提取、鹽析、離心后過膜直接供LC-MS/MS檢測分析。

圖3 不同凈化條件對蝦中農藥回收率的影響
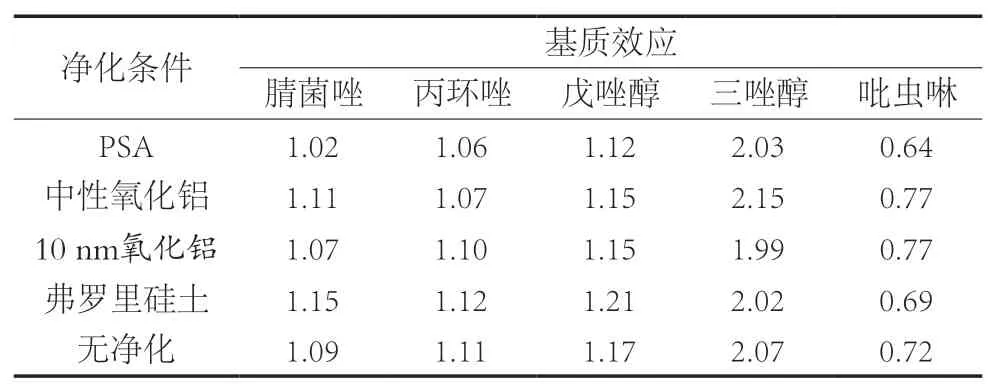
表2 不同凈化條件對農藥的基質效應的影響
2.2 分析方法的確證
2.2.1 溶劑標準曲線、基質標準曲線和基質效應
按照1.2.2的樣品前處理方法制得魚、蝦、白螺的空白樣品溶液,分別以空白樣品溶液和甲醇稀釋10 mg/L的5種農藥的混合標準溶液,配得質量濃度1.0,2.0,5.0,10,20,50,100和200 μg/L的基質標準溶液和溶劑標準溶液。采用LC-MS/MS檢測,以進樣濃度為橫坐標,農藥峰面積為縱坐標,分別繪制基質標準曲線和溶劑標準曲線,結果表明,魚、蝦、螺樣品基質中和純甲醇溶劑中5種農藥在1~200 μg/L的質量濃度范圍內均有良好的線性關系,基質標準曲線和溶劑標準曲線的相關系數(r)均在0.999 2~0.999 9范圍內。
按照式(1)計算農藥的基質效應,結果如表3所示。

表3 5種農藥的基質效應
當基質為蝦時,腈菌唑、丙環唑、戊唑醇無明顯的基質效應,三唑醇有明顯增強的基質效應,吡蟲啉有明顯減弱的基質效應;當基質為魚時,腈菌唑無明顯基質效應,丙環唑、戊唑醇和三唑醇表現為明顯減弱的基質效應,吡蟲啉為明顯增強的基質效應;當基質為白螺時,除三唑醇沒有明顯的基質效應外,其余4種均有明顯減弱的基質效應。
2.2.2 準確度和精密度

接表4
為驗證試驗方法的準確度和精密度,在蝦、魚、白螺3種水產品中進行5,20和50 μg/kg 3個質量分數下的添加回收率試驗,所得平均添加回收率和變異系數如表4所示。結果表明,5種農藥的平均添加回收率為70.55%~107.34%,C.V.為0.73%~10.35%,符合農藥殘留分析的要求。

表4 蝦、魚和白螺中5種農藥的平均添加回收率和相對標準偏差(n=6)
2.2.3 檢出限和定量限
按照3倍信噪比判斷檢出限,10倍信噪比判斷定量限,分別得到5種農藥的檢出限為1 μg/kg、定量限為5 μg/kg。
2.2.4 實際樣品測定
為檢驗試驗方法的可行性和實用性,從廣東湛江市場中隨機采購魚、蝦、螺各20個樣品進行檢測,圖4為質量濃度20 μg/L的蝦基質標準溶液和蝦的實際樣品的總離子流圖。60個魚、蝦、螺樣品均未檢出腈菌唑、丙環唑、戊唑醇、三唑醇和吡蟲啉5種農藥。

圖4 質量濃度20 μg/L的蝦基質標準溶液的總離子流圖(A)及蝦實際樣品的總離子流圖(B)
3 結論
試驗通過對提取溶劑、提取方式、鹽析條件、凈化條件等進行優化,建立LC-MS/MS快速檢測水產品中的腈菌唑、丙環唑、戊唑醇、三唑醇和吡蟲啉的殘留分析方法。試驗方法靈敏度高、可靠性和實用性良好,可作為水產品中農藥殘留快速定量分析的一種新方法。
