靶向免疫檢查點PD-1/PD-L1小分子PET探針的研究進展
2023-03-02 08:51:30代鵬飛綜述徐慧琴審校
安徽醫科大學學報 2023年2期
代鵬飛 綜述 徐慧琴 審校
近年來,免疫檢查點[程序性細胞死亡受體蛋白-1/程序性細胞死亡配體蛋白-1(programmed cell death protein-1/programmed cell death ligand protein-1,PD-1/PD-L1),細胞毒性T淋巴細胞相關蛋白-4(cytotoxic T-lymphocyte-associated protein- 4,CTLA- 4),淋巴細胞激活基因-3(lymphocyte activation gene-3,LAG-3)等]抑制劑是臨床研究最多和發展最快的免疫治療方法,尤其是PD-1/PD-L1免疫檢查點抑制劑受到了研究者廣泛關注[1-3]。由于惡性腫瘤在時間和空間存在高度異質性,免疫組織化學方法(immunohistochemistry,IHC)存在一定的局限性[4]。核醫學分子影像技術,尤其是PET,可以無創、精確、實時、動態、全面實現在體檢測PD-1/PD-L1表達水平,從而有效地篩選出能從PD-1/PD-L1免疫治療中獲益的患者。目前已發展的靶向PD-1/PD-L1免疫檢查點PET探針多數屬于單克隆抗體和抗體片段的PET探針[5]。然而,這些類型探針由于分子量大、代謝時間長、免疫原性、復雜的制備工藝限制了其在臨床上的應用。所以,開發小分子PET探針至關重要。基于目前少有文獻總結靶向免疫檢查點PD-1/PD-L1多肽和有機小分子PET探針的發展情況,本文主要總結該領域的研究進展,以期為發展新型的靶向PD-1/PD-L1小分子PET探針提供新的思路和帶來新的機遇。
1 PD-1/PD-L1信號通路概述
PD-1是含有268個氨基酸的誘導表達I型跨膜蛋白,屬于免疫球蛋白B7-CD28超家族成員,經誘導后主要表達于活化的T細胞、B細胞、調節性T細胞、NK細胞和樹突狀細胞等免疫細胞表面。PD-L1是含有290個氨基酸的I型跨膜蛋白,屬于免疫球蛋白B7超家族成員,主要表達于活化的T細胞、B細胞、抗原呈遞細胞(antigen presenting cell,APC)、巨噬細胞和多種癌細胞(黑色素瘤、乳腺癌、非小細胞肺癌、胃癌、膀胱癌、腎癌、卵巢癌等)表面。PD-1與配體PD-L1結合后,PD-1胞內區具有磷酸化作用位點C端氨基酸殘基的免疫受體酪氨酸轉換基序(immunoreceptor tyrosine-based switch motif,ITSM),在酪氨酸位點發生磷酸化,募集酪氨酸蛋白磷酸酶SHP-2,從而使下游的脾臟酪氨酸激酶(spleen tyrosine kinase,Syk)和磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)去磷酸化,進而抑制下游的AKT、mTOR、RAS、MEK、ERK等通路的活化,最終抑制T細胞活化、增殖以及細胞因子的分泌。在健康的生物機體內,這種免疫的負調控機制能使機體免于自身免疫應答過強出現的免疫損傷,避免了自身免疫性疾病(系統性紅斑狼瘡、類風濕性關節炎、胰島素依賴性糖尿病等)的發生。PD-L1在黑色素瘤、乳腺癌和非小細胞肺癌等癌細胞表面過度表達,從而持續異常地激活免疫檢查點PD-1/PD-L1信號通路,進而負性調節免疫應答系統,實現了癌細胞的免疫逃逸,造成了癌細胞的惡性增殖。阻斷免疫細胞表面PD-1與癌細胞表面PD-L1的結合,抑制PD-1/PD-L1信號通路的活化,阻斷負性調控信號,釋放活化T細胞的免疫應答能力,從而修復機體正常免疫反應并控制和清除癌細胞。因此,免疫檢查點PD-1/PD-L1信號通路是一個理想的免疫治療靶點[6]。
2 PD-1/PD-L1表達作為腫瘤免疫治療的重要生物標志物
迄今為止,全球已經有數十種抗體類PD-1/PD-L1免疫檢查點抑制劑獲批上市,用于黑色素瘤、結直腸癌、非小細胞肺癌、尿路上皮癌、腎細胞癌和肝癌等癌癥的治療[7]。多項臨床試驗證實,癌癥患者體內PD-1/PD-L1表達水平與其是否能從PD-1/PD-L1免疫檢查點抑制劑治療中獲益存在一定的正向相關[8]。目前,PD-1/PD-L1免疫檢查點抑制劑總體響應率僅為15%~40%,而且單獨使用PD-1單抗抑制劑的總體響應率僅為20%[9]。一項關于派姆單抗(pembrolizumab)用于非小細胞肺癌(NSCLC)的Ⅲ期臨床試驗表明[10],患者的客觀緩解率(objective response rate,ORR)、無進展生存期(progression-free survival,PFS)和中位生存期(median survival time,MST)與PD-L1表達水平呈正相關。并且,當患者體內PD-L1表達超過50%時,ORR達到45.2%。納武單抗(nivolumab)用于非小細胞肺鱗癌臨床試驗表明,PD-L1表達陽性患者的ORR和MST分別為31%和17.2個月,而PD-L1表達陰性患者的ORR和MST僅為9%和10.4個月。2020年NCCN發布的NSCLC指南中,明確指出了不同PD-1/PD-L1表達水平使用不同免疫治療方案。所以,準確檢測PD-1/PD-L1表達水平,可以有效地篩選能從PD-1/PD-L1免疫治療中獲益的患者,對患者進行分層治療、療效監測和預后評價具有重要意義。
3 靶向免疫檢查點PD-1/PD-L1抗體類PET探針
近年來,靶向免疫檢查點PD-1/PD-L1的單克隆抗體、雙特異性抗體、納米抗體、抗體片段、單鏈抗體、親合體、Adnectin和類抗體支架蛋白等的PET分子探針到了迅速發展[11-13],本文概括了一些靶向免疫檢查點PD-1/PD-L1抗體類PET探針的情況,見表1。雖然免疫PET顯像(Immuno-PET)發展迅速、特異性好,但是相對分子量大、生物半衰期長、免疫原性強、制備困難、需要特殊的基因組編輯技術、肝臟非特異性高攝取等缺陷,使其具有一定的局限性。
4 靶向免疫檢查點PD-1/PD-L1小分子PET探針
相比于單克隆抗體、雙特異性抗體、納米抗體、抗體片段、單鏈抗體、親合體和Adnectin等類抗體支架蛋白,小分子多肽和有機小分子化合物具有以下優點:生物體內半衰期短、組織穿透力強、可修飾性強、無免疫原性、合成簡單、成本廉價、化學穩定性好、運輸便捷和顯像時間短等優點[24]。
4.1 核素標記的多肽小分子PET探針WL12是一種由12個氨基酸構成的高親和性靶向PD-L1環狀肽,其半數抑制濃度(half inhibit concentration, IC50)值為20 nmol/L[25]。分子對接相互作用分析表明,WL12在PD-L1的凹槽中形成了β片狀結構。WL12的D-亮氨酸類似于PD-1的Ile134插入到疏水性口袋中,其中一個正亮氨酸殘基結合方式與PD-1的Ile126結合方式相同。此外,WL12與PD-L1之間存在大量的氫鍵。有研究者在放射性核素標記多肽小分子抑制劑WL12制備多肽小分子PET探針領域做了大量的工作[26-28],見表2。
2017年,有研究者將DOTAGA雙功能螯合劑連接在WL12多肽上[26]。隨后,使用64Cu進行放射性核素標記制備了多肽小分子PET探針[64Cu]WL12。研究顯示[64Cu]WL12與PD-L1具有較強親和力(Kd=2.9 nmol/L)。隨后在構建的PD-L1表達陽性和PD-L1表達陰性的中國倉鼠卵巢細胞(CHO)腫瘤的荷瘤鼠模型中進行PET-CT顯像,研究表明,在注射10 min后即可在PD-L1表達陽性的荷瘤鼠模型中觀察到[64Cu]WL12的高攝取,可以持續到120 h,免疫組化證實了該結果;而PD-L1表達陰性的荷瘤鼠模型中并未觀察到[64Cu]WL12的高攝取。hPD-L1腫瘤的腫瘤-肌肉和腫瘤-血液比值分別為25.6±1.9和4.7±1。結果表明[64Cu]WL12在注射1 h后,可以特異性檢測腫瘤PD-L1的表達水平。
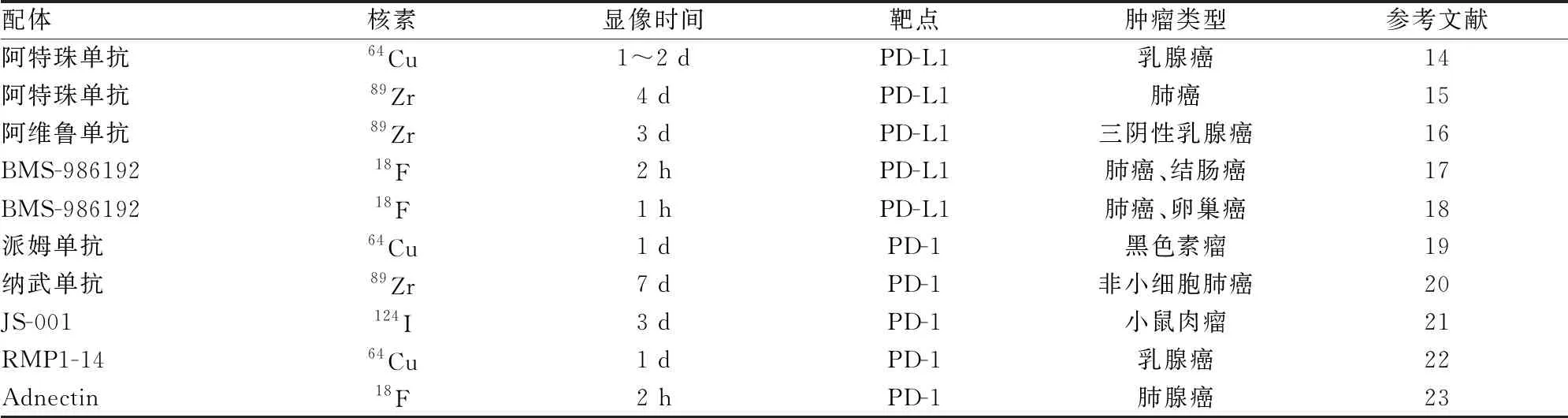
表1 靶向免疫檢查點PD-1/PD-L1抗體類PET探針
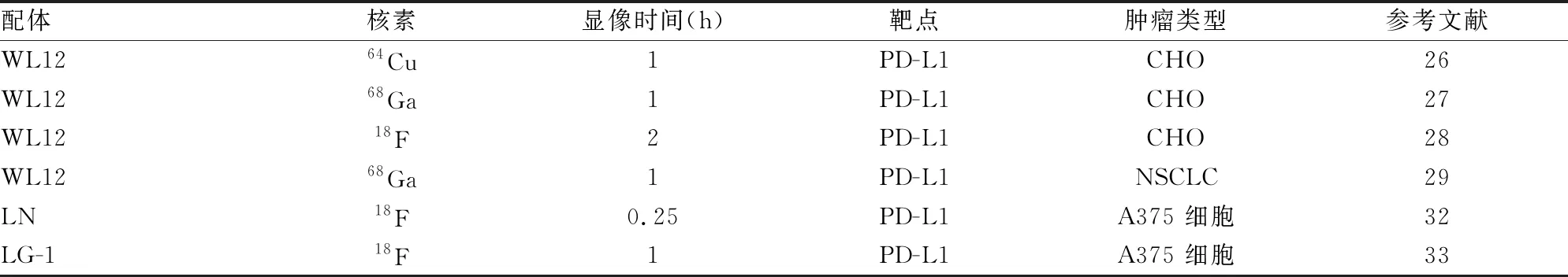
表2 靶向免疫檢查點PD-1/PD-L1小分子PET探針
68Ca的半衰期與WL12多肽的生物半衰期相近,并且68Ga方便獲取(68Ge/68Ga發生器),方便臨床工作人員的使用。2018年,De Silva et al[27]用放射性核素68Ca標記DOTAGA-WL12多肽制備了68Ca 標記的多肽小分子PET探針[68Ca]WL12。生物分布實驗表明,在所有不同時間點的測試中,[68Ca]WL12在hPD-L1腫瘤中的攝取值是對照組的9倍。在注射15、60、120 min時,hPD-L1腫瘤的每克組織百分注射劑量率(%ID/g)分別為19.4±3.3、11.56±3.18、9.89±1.72;而對照組腫瘤在注射15、60、120 min時,%ID/g均小于1.33±0.21。相比于[64Cu]WL12,[68Ca]WL12制備方便、具有更適于臨床應用的生物半衰期。
18F來源于回旋加速器,實用性更強,更適合于PET成像特性。2019年,Lesniak et al[28]使用[18F]FPy-TFP作為放射性輔基通過與WL12鳥氨酸側鏈氨基形成酰胺鍵,從而制備了18F標記的多肽小分子PET探針[18F]FPy-WL12。PET-CT圖像顯示,在放射性示蹤劑注射10、30和60 min后,hPD-L1腫瘤中[18F]FPy-WL12的攝取值與對照組CHO相比顯著增加。在注射10、60、120 min時,hPD-L1腫瘤的%ID/g分別為5.23±1.11、7.16±1.67、8.86±10.2;而對照組腫瘤%ID/g均小于1.77±0.21。然而研究發現肝臟和腎臟對[18F]FPy-WL12攝取率較高。
2022年,Zhou et al[29]利用68Ca標記的多肽小分子WL12制備了PET探針68Ga-NOTA-WL12,同時進行了人體研究,結果表明,在體外和體內PD-L1表達陽性腫瘤中,均表現特異性攝取68Ga-NOTA-WL12;生物分布實驗表明,68Ga-NOTA-WL12主要分布在肝、脾、小腸和腎;在注射1 h后,腫瘤清晰可見,尤其是肺,1 h時腫瘤/肺比值為4.45±1.89。
4.2 核素標記的有機小分子PET探針近年來,隨著小分子抑制劑與PD-1/PD-L1蛋白作用模式的研究,越來越多的分子(磺酰胺類、聯苯類、噁二唑類、芐苯醚類等)已經用于抑制免疫檢查點PD-1/PD-L1的結合[30]。
2015年,美國百時美施貴寶公司在申請的專利中披露了具有抑制PD-1/PD-L1結合活性的聯苯類化合物[31]。受此啟發,2020年,Miao et al[32]合成了結構上類似于BMS-1166的化合物LN2。通過銅催化的炔基和疊氮的點擊反應(Husigen環加成反應)將硼氨酸引入到化合物LN2中,制備成含硼氨酸LN。最后,基于18F-19F同位素交換制備成有機小分子PET探針[18F]LN。實驗結果表明,[18F]LN與PD-L1具有較強親和力(Kd = 65.27±3.47 nmol/L)。同時在建立的PD-L1陽性(A375-hPD-L1)和PD-L1陰性(A375)的荷瘤鼠模型中顯像。結果表明,在注射15 min后,在PD-L1陽性(A375-hPD-L1)的荷瘤鼠腫瘤攝取值為(1.96±0.27)%ID/g;在PD-L1陰性(A375)和阻斷組的荷瘤鼠腫瘤攝取值分別為(0.89±0.31)%ID/g和(1.07±0.26)%ID/g。研究分析,通過點擊反應引入親脂性的AMBF3降低了探針對PD-L1的親和力,從而造成非靶器官的非特異性攝取。
為了改善探針的親水性,2021年,Lv et al[33]基于醛和羥胺縮合策略,使用[18F]FDG代替AMBF3作為標記輔基標記有機小分子L7制備了[18F]LG-1。[18F]FDG結構的引入顯著提高了[18F]LG-1的水溶性,其Log Do/w比[18F]LN低4.8倍。體外實驗表明,PD-L1表達陽性細胞(A375-hPD-L1)攝取明顯高于PD-L1表達陰性細胞(A375)。體內動態PET圖像表明,在注射60 min后,在PD-L1陽性(A375-hPD-L1)的荷瘤鼠腫瘤攝取值是PD-L1陰性(A375)荷瘤鼠腫瘤的2.6倍。總之,[18F]LG-1是一種潛在的靶向PD-L1的有機小分子PET探針。
5 總結和展望
研究[4]表明有創的IHC在檢測PD-1/PD-L1表達水平方面存在局限性:① 檢測閾值不同;② 容易受到干擾素、淋巴細胞趨化因子和前期治療等因素影響;③ 無法實時、重復和動態監測PD-1/PD-L1表達。靶向PD-1/PD-L1免疫檢查點PET探針可以高效地實現活體檢測PD-1/PD-L1表達水平。相比于抗體類PET探針,小分子PET探針具有生物體內半衰期短、化學穩定性好、無免疫原性和成本廉價等優點。然而,PD-1與PD-L1接觸的界面具有高度平坦且疏水的結構,沒有小分子結合的合適位點,導致PD-1/PD-L1免疫檢查點小分子抑制劑研究進展緩慢。由于缺乏高活性的PD-1/PD-L1小分子抑制劑,使得靶向免疫檢查點PD-1/PD-L1小分子PET探針的研究進展更加緩慢。
