葛根素抑制內質網應激減輕壓力負荷引起小鼠心肌肥厚實驗研究
2023-02-17 01:31:06劉艷麗董文婷尚福軍馮海濤
陜西醫學雜志 2023年2期
劉艷麗,董文婷,尚福軍,馮海濤,郭 錦,王 錦
(1.西安國際醫學中心醫院,陜西 西安 710100;2.西安市臨潼區婦幼保健院,陜西 西安710600)
心肌肥厚是心臟受到各種刺激時,為了維持心輸出量,出現的一種適應性和代償性反應[1]。由于成熟心肌細胞已經成為一種終末分化細胞,幾乎喪失分裂和增殖能力,只能通過肥厚性重塑改變其體積和重量以增加心肌收縮力[2]。然而,長期的慢性刺激如高血壓會導致心臟失代償而進展為病理性心肌肥厚,并最終導致心臟功能障礙和心力衰竭。臨床研究[3-4]表明,心肌肥厚是觸發心力衰竭發生不良后果和心血管死亡事件增加的一個獨立危險因素,會導致擴張型心肌病、缺血性心臟病甚至猝死。心肌肥厚是一個非常復雜的病理重塑過程,伴有心肌纖維化、細胞凋亡和心臟功能障礙[5-6]。然而,臨床上缺乏防治病理性心肌肥厚的藥物。葛根素(Puerarin,Pue)是黃酮類化合物,化學名為7,4’-二羥基異黃酮-8β-吡喃葡萄糖苷,是從中藥葛根中提取的主要活性成分[7]。葛根素的藥理作用包括改善微循環、降低炎性反應和細胞凋亡、清除氧自由基和改善胰島素抵抗,使其成為治療高血壓、腦缺血、心肌缺血、糖尿病和動脈硬化的潛在藥物[8-9]。但是Pue能否通過內質網應激調節心肌肥厚尚不清楚。本實驗將通過腹主動脈縮窄(Abdominal aortic constriction,AAC)方法建立心肌肥厚動物模型,觀察Pue灌胃給藥4周后對壓力負荷引起的心肌肥大及對內質網應激的調控作用。
1 材料與方法
1.1 實驗動物與試劑 在西安交通大學實驗動物中心購買60只8周齡成年雄性C57/bl小鼠;Pue(P5555)購買于Sigma公司;RNA提取試劑盒和反轉錄試劑盒(FP209)購于天根生化科技(北京)有限公司;PERK(3192S)、CHOP(2895S)和GRP78(3183S)抗體均購自Cell Siginal Technology公司;GAPDH(60004-1-Ig)、辣根過氧化物酶標記的山羊抗鼠(PR30012)和山羊抗兔(PR30011)二抗購自武漢三鷹生物技術有限公司。
1.2 心肌肥厚模型的制備 待雄性C57/bl小鼠麻醉后,將小鼠固定于小動物手術操作臺上,行氣管插管并連接呼吸機,采用AAC引起心肌肥厚。方法如下:無菌條件下通過腹部中線切口暴露腹主動脈,并沿腹主動脈放置一根18號針。在腹主動脈和腎動脈上方的針頭周圍緊密地綁著一條結扎線(7-0絲線)。隨后取下針頭,形成縮窄區。假手術組小鼠手術操作與模型組相同,但不結扎腹主動脈。手術完成后,逐層縫合,并將小鼠放在暖墊上,待到它們從麻醉中恢復。
1.3 實驗分組 60只小鼠隨機分成四組(n=15):假手術組(Sham組);Pue對照組(Pue組);腹主動脈縮窄術組(AAC組);Pue干預AAC組(Pue+AAC組)。根據以往文獻[10]報道,Pue組和Pue+AAC組術后腹腔注射50 mg/(kg·d) Pue,持續4周,Sham組和AAC組腹腔注射等量的0.9%氯化鈉溶液,持續4周,所有小鼠均采用正常飲食,可自由攝取水和食物。
1.4 小動物超聲檢測心臟功能 采用小動物超聲儀檢測心臟功能,AAC術后4周,小鼠麻醉后采用脫毛膏去除胸前區被毛,用VEVO 770小動物超聲儀檢測左室后壁厚度(Left ventricular posterior wall thickness,LVPWd)、左室內徑(Left ventricular diameter,LVId)和室間隔厚度(Interventricular septal depth,IVSd)。
1.5 RT-PCR檢測ANP和Myh7 mRNA水平 根據RNA提取試劑盒步驟提取心臟組織總RNA,采用分光光度儀檢測RNA濃度,并用反轉錄試劑盒生成cDNA;配制總體積為20 μl的反應體系,其中包括:cDNA 2 μl、超純水7.4 μl、引物0.8 μl和SYBR Green Mix 9 μl。擴增程序設為:95 ℃預變性10 min,95 ℃變性10 s,60 ℃退火15 s,72 ℃延伸10 s,共38個循環。計算Ct值、2-ΔΔCt值。以GAPDH作為內參,引物序列見表1。

表1 PCR擴增引物序列
1.6 心臟重量與體重比值 實驗結束后,將小鼠麻醉稱重并記錄每只小鼠體重(Body weight,BW),然后通過頸動脈放血將小鼠處死,快速開胸取下心臟并稱重,記錄心臟重量(Heart weight,HW),計算心臟重量/體重比值(HW/BW)。
1.7 Masson染色 實驗結束后,將小鼠麻醉,快速開胸取出心臟,在預冷的PBS中沖洗,然后放入4%多聚甲醛中固定,行常規二甲苯和梯度乙醇脫水,石蠟包埋,并切成3 μm厚的薄片。行常規Masson染色方法步驟,染好后用中性樹脂封片,在普通光學顯微鏡下拍照,Image J軟件計算纖維化面積比例。
1.8 Western blot檢測PERK、GRP78和CHOP蛋白的表達 常規方法提取心臟組織蛋白,BCA法定量蛋白濃度;配制SDS-PAGE凝膠,每孔蛋白樣品上樣量相同,恒壓90 V電泳100 min,濕轉法恒流200 mA轉膜90 min;將轉好的PVDF膜用5%脫脂奶粉室溫封閉90 min;根據分子量大小將目的條帶切下分別放入PERK(1∶1000)、GRP78(1∶1000)、CHOP(1∶1000)和GAPDH(1∶5000)抗體管4 ℃搖床孵育12 h;TBST洗膜3次,10 min/次,用辣根過氧化物酶標記的山羊抗兔(1∶5000)和山羊抗鼠二抗(1∶5000)室溫搖床孵育120 min,TBST洗膜3次,10 min/次,ECL顯色,Image J軟件進行灰度值分析。
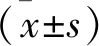
2 結 果
2.1 Pue改善AAC引起的心臟室壁厚度增加 見圖1。和Sham組比較,Pue組LVPWd、IVSd和LVId值無統計學差異(均P>0.05);和Sham組比較,AAC組LVPWd和IVSd值增加,LVId降低(均P<0.01);和AAC組比較,AAC+Pue組LVPWd和IVSd值降低,LVId增加(均P<0.01)。

注:與Sham組比較,*P<0.01;與AAC組比較,#P<0.01
2.2 Pue抑制AAC引起的HW/BW值、ANP和Myh7 mRNA水平增加 見圖2。和Sham組比較,Pue組HW/BW值、ANP和Myh7 mRNA水平均無統計學差異(均P>0.05);和Sham組比較,AAC組HW/BW值、ANP和Myh7 mRNA水平增加(均P<0.01);和AAC組比較,AAC+Pue組HW/BW值、ANP和Myh7 mRNA水平降低(均P<0.01)。

注:與Sham組比較,*P<0.01;與AAC組比較,#P<0.01
2.3 Pue抑制AAC引起的心肌細胞增大 見圖3。WGA染色結果顯示Sham組和Pue組心肌細胞橫截面積比較無統計學差異(均P>0.05);和Sham組比較,AAC組心肌細胞橫截面積明顯增加,和AAC組比較,AAC+Pue組心肌細胞橫截面積減小(均P<0.01)。
2.4 Pue抑制AAC引起的心肌纖維化 見圖4。Masson染色結果顯示Sham組和Pue組心肌纖維化程度比較無統計學差異(均P<0.01);和Sham組比較,AAC組心肌纖維化程度明顯增加(P<0.01);和AAC組比較,AAC+Pue組心肌纖維化程度降低(P<0.01)。
2.5 Pue抑制AAC引起的內質網應激 見圖5。和Sham組比較,Pue組內質網應激相關蛋白PERK、GRP78和CHOP蛋白表達均無統計學差異(均P>0.05);和Sham組比較,AAC組PERK和CHOP蛋白表達明顯增加,GRP78表達降低(均P<0.01);和AAC組比較,AAC+Pue組AAC組PERK和CHOP蛋白表達明顯減少,GRP78表達增加(均P<0.01)。

注:與Sham組比較,*P<0.01;與AAC組比較,#P<0.01

注:與Sham組比較,*P<0.01;與AAC組比較,#P<0.01

注:與Sham組比較,*P<0.01;與AAC組比較,#P<0.01
3 討 論
心肌肥厚是心臟對生理或病理性刺激的一種代償性反應機制,成年心肌細胞沒有增殖能力,只能通過單個細胞面積和體積增大,增加心室肌壁厚度和質量,進而維持心肌收縮力、心輸出量和全身循環血量的需求[10]。心肌肥厚涉及多種復雜的機制,包括血流動力學、神經體液激素激活、生長因子/細胞因子、缺氧、衰老和糖尿病等一系列的變化,表現為心肌細胞質量增加、肌節重排和心肌間質細胞外基質沉積。流行病學研究表明,心肌肥厚是心血管病發病和死亡的獨立危險因素[11]。闡明心肌肥厚的病理機制并尋找潛在的防治方法成為研究的重要方向。
Pue作為傳統中藥葛根中提取的主要生物活性成分,在心血管疾病如缺血再灌注、心肌梗死、糖尿病心肌病和動脈粥樣硬化等方面均具有較好的治療效果,在臨床上得到了廣泛應用,其作用機制與抑制細胞凋亡、降低氧化應激、減輕炎性反應有關[12-14]。Pue在心肌肥厚中的作用也得到了廣泛關注,有研究報道,在血管緊張素Ⅱ誘導的小鼠心肌肥厚模型中,Pue可通過上調miR-15b/195抑制TGF-β信號通路減輕心肌肥厚[15];Pue還能激活Nrf2并調控ROS含量,抑制氧化應激損傷和心肌肥厚[16];此外Pue還可以通過激活Sirt1信號改善心肌缺血再灌注損傷[8],但Pue在AAC引起的心肌肥厚中的作用和機制還未見報道。本實驗發現,Pue干預AAC能明顯減小LVPWd和IVSd值,增加LVId,Pue可以降低AAC引起的HW/BW值、ANP和Myh7 mRNA水平增加;心肌肥厚會導致心肌細胞橫截面積增大、細胞外基質重塑引起心肌纖維化加重。本實驗發現,AAC引起的心肌肥厚中心肌細胞橫截面積和纖維化明顯增加,而Pue干預AAC可明顯降低心肌細胞橫截面積和纖維化,這些結果表明,Pue能夠改善AAC引起的心肌肥厚。
內質網是細胞內一種重要的細胞器,在蛋白質合成、折疊和轉運,鈣穩態的調控以及脂質合成中均發揮關鍵作用[17-19]。在某些病理因素的刺激下,內質網功能穩態被破壞,導致錯誤折疊和未折疊蛋白質的積累,進而觸發內質網應激[20]。內質網應激通過影響轉錄和翻譯,激活未折疊蛋白反應以恢復內質網功能的穩態。然而,過度的內質網應激會導致細胞功能障礙并誘發細胞凋亡。研究表明,內質網應激參與各種心臟病的發展和進展,如心肌肥大、缺血性心臟病和心力衰竭[21]。本實驗發現,AAC明顯增加內質網應激蛋白PERK和CHOP的表達,降低GRP78表達;而Pue干預AAC后抑制PERK和CHOP的表達,增加GRP78的表達。
綜上所述,本實驗結果說明,Pue能通過抑制心肌纖維化改善AAC引起的心肌肥厚,其作用機制與減輕內質網應激有關。這些結果為臨床上進一步探討Pue治療心肌肥厚提供了實驗依據。然而,這項研究仍存在一定的不足,由于只觀察了心肌肥厚時Pue對內質網應激的作用,而沒有探討Pue如何調控內質網應激,因此,應當進一步闡明Pue調控內質網應激的信號通路。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
學苑創造·A版(2020年9期)2020-10-13 09:41:02
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
七彩語文·畫刊(2012年3期)2012-04-29 00:00:00
