OLED發光材料的理論計算與分子設計
2023-02-16 10:51:32劉美惠
發光學報 2023年1期
劉美惠, 彭 謙
(中國科學院大學 化學科學學院, 北京 100049)
1 引 言
有機發光二極管(OLED)在柔性顯示、固態照明、有機激光、化學/生物傳感和光通信等高科技領域具有廣闊的應用前景,其發展備受科學界和工業界的關注[1-7]。OLED器件獨特的優勢很大程度上來源于有機發光材料的多樣性、可塑性及可設計性。根據OLED器件的工作原理[8-9](圖1)可知,器件的外量子效率(EQE)由4個因素決定ηexe=ηrecηexcηplηoc。其中ηrec是載流子形成束縛對的效率,主要取決于半導體中載流子的注入和傳輸性質;ηexc是發光激子的生成比率,根據自旋量子統計,單線態激子占電生激子總數的25%,三線態激子占75%;ηpl是光致發光量子效率(PLQY);ηoc是光學耦合常數,即產生的全部光子從器件中出射的比例。由該公式可知,在不考慮光取出的情況下,提高發光材料的ηexc和ηpl是提升器件效率的關鍵。根據激子的利用方式及發展時間進行分類,第一代是傳統熒光材料(單線態發光),代表分子如三(8-羥基喹啉)鋁(Alq3)[10],受自旋統計的限制,激子利用率在理論上不會超過 25%,因此其器件的外量子效率一般不超過5%;第二代是過渡金屬配合物磷光材料(三線態發光)[11],以銥(Ir)和鉑(Pt)配合物為代表[12-13],能夠依靠重原子效應帶來的強自旋軌道耦合(SOC),實現 S1→T1以及 T1→S0的快速系間竄越(ISC)和輻射過程,從而使得激子利用率達到理論值100%,但卻存在造價昂貴、色度不全等問題。熱活化延遲熒光(TADF)材料是利用反系間竄越(rISC)將三線態轉化為單線態,然后單線態發光,其激子利用率理論上可達100%,成為第三代OLED材料,引起了人們的廣泛關注[14-15]。隨著OLED材料和器件的發展,人們提出了很多提高激子利用率的新方法和新材料,例如TADF主體材料[16]、單線態裂分主體材料[17]、單線態裂分-熱激子主體材料[18]、自由基發光材料[19]、熱激子-聚集誘導發光材料[20]、純有機室溫磷光材料[21-23]等。中國科學家在這些方面均做了許多原創性工作,為發展具有自主知識產權的市場化發光材料打下基礎。

圖1 OLED工作原理示意圖,包括(1)電荷注入、(2)電荷傳輸、(3)電荷復合和(4)激子衰減四個步驟。Fig.1 Schematic illustration of working principle OLED,including(1)charge injection,(2)charge transport,(3)charge re?combination and (4)exciton decays.
一旦激子形成之后,有機分子的發光量子效率取決于激子不同衰減途徑之間的競爭。如圖2所示,從激發態(單線態或三線態)到基態有三條衰減途徑:(ⅰ) 輻射過程(kr)[24];(ⅱ)在諧振區,由振動弛豫誘導的無輻射衰減過程(NR-VR,kVnrR)[25];(ⅲ)在非諧振區,通過最小能量交叉點的無輻射衰減過程(NR-MECP,kMnrECP)[26]。兩種無輻射衰減過程有可能同時存在,并與輻射過程產生競爭。這時,發光量子效率可表示為ηpl=kr(/kr+kVnrR+kMnrECP)。對于有機分子來說,結構比較柔性,發光過程必定伴隨著結構變化;但是,是NR-VR還是NR-MECP主導著無輻射過程,長期以來尚未有定論[27-28]。在NR-VR過程中,分子的幾何結構變化相對較小,其速率可以在諧振子模型下進行求解。在NRMECP過程中,分子的幾何結構變化相對較大,或者像圖2所示經過一個過渡態、局部最小點之后上升到MECP點,或者越過一個能壘衰減到MECP點,或者無需能壘衰減到MECP點。對一個實際體系,勢能面形狀和能壘的高低決定著哪個過程是決速步。從動力學理論的角度來看,無論無輻射衰減是VR還是MECP引起的,非絕熱動力學原則上可以給出準確結果。但是,目前非絕熱動力學模擬方法的模擬時間是飛秒或皮秒級[29],遠小于輻射衰減的納秒級。這里,我們對于諧振區域采用熱振動關聯函數(TVCF)速率理論[30-31],對于非諧區域采用過渡態理論,對熒光[32]、磷光[33]、熱活化延遲熒光[34-35]等各種發光材料的激發態衰減速率和發光量子效率進行定量計算,揭示其機理,建立結構-性能關系,提出有效的描述符和分子設計原則,設計出優良性能的發光材料。

圖 2 (a)從激發態(ES)到基態(GS)的衰減路徑示意圖[32]:(ⅰ) 輻射過程(k)r;(ⅱ)振動弛豫誘導的無輻射衰減(NR-VR,);(ⅲ)最小能量交叉點誘導的無輻射衰減(NR-MECP);(b)PCM和QM/MM的計算模型[36(] 以分子40為例)。Fig.2 (a)Schematic graph of the decay pathways from an excited state(ES) to the ground state( GS)[32]:( ⅰ) radiative decay(kr),(ⅱ) the nonradiative decay via vibration relaxation in harmonic region( NR-VR,),and( ⅲ) the nonradiative decay via MECP( NR-MECP,) beyond harmonic region.( b)Computational setups of PCM and QM/MM[36]( taking molecule 40 as an example).
2 理論與計算方法
基于費米黃金規則和一階微擾理論,振動弛豫誘導的無輻射速率常數knr可以寫作[37-38]:

其中H′表示兩個態的相互作用,包括HBO非絕熱耦合和HSO自旋軌道耦合;r和Q分別是電子和核正則坐標;Φ和Θ分別是電子和振動波函數;νi、νf分別是初始電子態i和末電子態f的振動態。
多重度相同的電子態之間的躍遷被稱為內轉換,主要由非絕熱耦合引起,則有
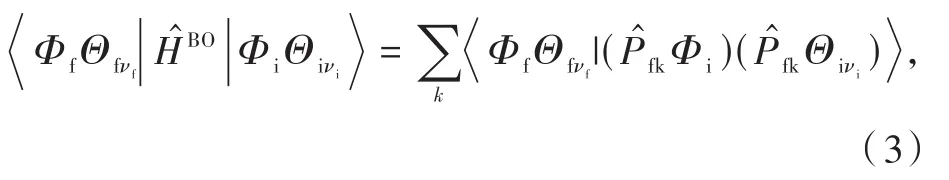
其中,是核動量算子,這里忽略小項?2Φi/?Q2fl。在諧振子模型和Condon近似下,振動弛豫誘導的內轉換速率kVICR可以表示為熱振動關聯函數的形式:

同理,多重度不同的電子態之間的系間竄越速率常數也可以寫為熱振動關聯函數的形式:

有限溫度下,自發輻射速率常數是對發射光譜在全波段范圍內的積分:
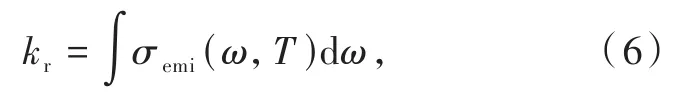
其中,發射光譜:
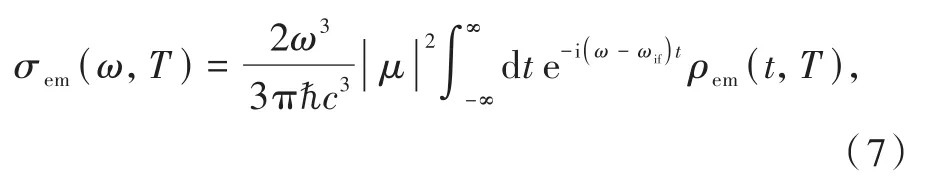
μ是躍遷偶極矩矩陣元,ρem(t,T)與ρISC(t,T)形式相同,求解方法相同。


其中,ΔG≠表示在3ES和3MECP之間的吉布斯自由能。
利用以上理論公式,結合量子化學,定量計算各種發光材料在不同環境下的發光量子效率。這里,分別采用連續極化模型(PCM)和量子力學/分子力學方法(QM/MM)來考慮溶液和聚集環境。量化計算單個分子激發態采用TD-DFT方法。在本文中,我們選擇兩種方法進行QM/MM計算,一種是使用CHEMSHELL 3.5接口程序[40],其中的QM計算使用 TURBOMOLE 6.5程序包[41],MM使用DL_POLY程序包[42];另一種是使用Gaussian 16中的ONIOM方法。SOC通過PySOC軟件包[43]或者ORCA程序包[44]計算。MECP的優化在sobMECP程序包中完成[43]。輻射與無輻射速率常數通過MO?MAP 程序包計算[45]。
3 發光機理與分子設計
3.1 熒光體系
熒光材料因其成本低、穩定、色純度高仍在OLED材料中占有一席之地,特別是藍光發光材料。由OLED工作原理可知,有機分子通常在聚集態和固態下發光。相較于溶液中的“自由”分子,處于“限域”環境下的分子可以表現出完全不同的光響應行為。例如,在溶液中強發光體系的剛性共軛平面分子,在聚集態下會發生熒光猝滅;而在溶液中無發光或者弱發光的柔性體系,在聚集狀態下發出強光。我們結合量化方法,應用TVCF速率理論,定量計算了各種熒光體系(圖3)在氣相、溶液和固態等不同環境下的熒光量子效率,并與實驗測量值進行了比較(圖4(a))。這里,ηVRF=kr(kr+kVRnr),只 考 慮 了 輻 射 衰 減 和 振動弛豫誘導的無輻射躍遷。由圖4(a)可以看出,計算值與實驗值吻合很好,這說明:(1)量化方法和TVCF方法對振動弛豫誘導的無輻射速率的描述是可靠的;(2)在這些體系中,忽略MECP誘導的無輻射衰減是合理的。
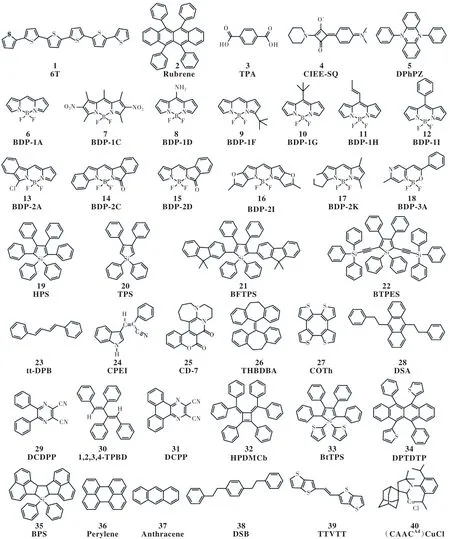
圖3 熒光分子的分子結構[32]Fig.3 Chemical structures of investigated fluorescent compounds[32]
由圖 4(a)可以看到,分子 19、27、32及 40在溶液中的熒光量子效率很低,而在聚集狀態下提高,表現出聚集誘導或者增強發光現象。經過對比分析兩種環境下的激發態性質和衰減速率常數,我們發現,分子聚集可以削弱電子與多種不同類型的振動(包括環旋轉[46]、鍵彎曲[47]和鍵搖擺[48])之間的耦合及振動-振動之間的耦合(圖4(b)),急劇減慢無輻射躍遷,使得輻射躍遷占主導,致使熒光增強。電子-振動耦合/振動-振動耦合對環境的敏感性可以通過實驗可測的瞬態共振拉曼光譜和同位素效應進行驗證[25,49]。最重要的是,我們可以將電子-振動耦合投影到分子內坐標上,能夠清晰地勾勒出電子-振動耦合和振動-振動耦合所在的化學結構片段,以此提出了分子化學結構的片段改造、移除、重組及改變分子的聚集結構和周圍環境等手段來達到解耦合的化學結構策略,有助于設計高效發光的固態熒光體系[32]。

圖4 (a)有機體系在溶液和固相下的理論計算與實驗測量的發光量子效率ηVF R =kr (kr+)的對比;(b)NR-VR通道中起主要貢獻的振動模式示意圖[32]。Fig.4 (a)Comparison between experimentally measured and the theoretically calculated (ηVF R =kr (kr+) luminescence quantum efficiency for the organic systems in solution and solid phase.( b)Illustrations of vibrational modes involved in the NR-VR channels with dominant contribution to reorganization energy[32].
3.2 磷光體系
金屬有機配合物磷光材料由于高的激子利用率成為第二代 OLED發光材料。其中,Pt(Ⅱ)配合物因具有短壽命和高效率而備受關注[50-51]。目前,商業化的磷光材料僅限于綠光和紅光,藍光器件的穩定性和效率滾降問題尚未得到有效解決。所以,高效的深藍光磷光材料亟待研發。最近,對藍光Pt(Ⅱ)配合物的研究發展迅猛,其磷光效率已經接近100%,并且磷光壽命在室溫下是微妙級別[52-53]。Pt(Ⅱ)配合物具有二齒配體、三齒配體、四齒配體等多種配體結構,表現出豐富的化學結構和光物理性質[54-55]。而且,在四齒配體中,不同的環金屬配體也能形成不同的平面或者扭曲結構,導致迥異的發光性質[56-57]。但是,Pt(Ⅱ)配合物具有不飽和的d軌道,容易形成金屬中心(3MC)態,猝滅發光。為了考查Pt(Ⅱ)配合物的結構-性能關系,我們選擇16個5/6/6金屬環四齒配體Pt(Ⅱ)配合物(圖5)[58-61],應用TVCF和過渡態理論,系統地研究了其磷光輻射速率、振動弛豫誘導的無輻射速率和MECP誘導的無輻射速率及磷光量子效率[33]。
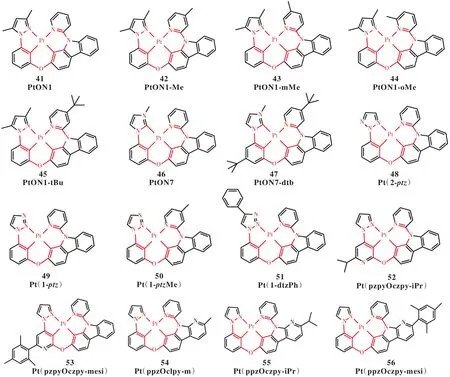
圖 5 四齒配體Pt(Ⅱ)配合物的分子結構Fig.5 Chemical structures of tetradentate Pt(Ⅱ) complexes
圖6(a)比較了計算和實驗的磷光量子效率ηP,其 中 ,ηVRP=kr/(kr+kVRnr),ηtotP=kr/(kr+kVRnr+kMECPnr)。由圖 6(a)可知,分子 44 的ηVRP偏離實驗值。分子44具有能壘較低的3MC態和MECP,其余分子的ηVRP與ηtotP幾乎重疊,并與實驗值高度吻合。這表明它們具有難以到達的3MC態和MECP,同時證明了TVCF速率理論定量預測Pt(Ⅱ)配合物的磷光量子效率的可靠性。圖6(b)是分子44的勢能面,穩定態3ES沿著Pt—N配位鍵的斷裂方向,越過較小能壘(Ea=0.41 eV,Eb=0.01 eV,Ec=0.30 eV)的3TS到達3MC態,之后借助于溫度效應熱分布到MECP,快速回到S0。而且,根據公式(8)和公式(9),由于分子44具有較小的吉布斯自由能,ka達到1010s-1,得到kMECPnr(8.06×105s-1)與kTVCFnr(4.75×105s-1)形成競爭關系。相較于其他體系,在Pt—N的鄰位上引入的兩個—CH3的位阻效應活化了Pt—N配位鍵,導致3MC生成。從圖6(a)還可以看出,沒有3MC生成,分子54~56的磷光效率也比較低。我們進一步分析了決定結構弛豫誘導的無輻射躍遷速率的重要參數重整能(圖6(c))。分子54~56的重整能遠大于其他體系的重整能。再從重整能對內坐標的投影結果來看(圖6(d)),增加的重整能主要來源于咔唑,柔性取代基的引入活化了咔唑。
總之,通過TVCF和過渡態理論,定量預測了藍光四齒配體Pt(Ⅱ)配合物的磷光量子效率,建立了結構-性能關系,為設計出高效的藍光Pt(Ⅱ)配合物提供了思路。例如,移除Pt—N的鄰位的取代基,避免3MC的生成;移除咔唑外圍上的柔性基團,避免大的結構弛豫引起的無輻射躍遷。
3.3 熱活化延遲熒光體系
TADF體系由于高穩定性和高激子利用率成為第三代OLED發光材料,具有很大的商業潛力[62-64]。在TADF機理中,電注入形成的三線態激子通過rISC過程轉化為單線態激子,從而發射熒光。所以,TADF是否發生取決于rISC的速率。人們公認的就是減少單線態-三線態能隙(ΔEST),增加SOC,來提高rISC速率。由此可知,在保證ΔEST足夠小的條件下,引入重原子會大大增強SOC,加快 rISC(108~1010s?1)速率并提高 TADF 效率[65-67]。但是,最近的實驗報道的兩個具有TADF性質的Au(Ⅲ)配合物[68-69(]如圖7(a)的57和58),配 合 物 57 的~ 1010s?1,TADF 效 率(ηTADF)為79%;配合物58的~107s?1而ηTADF高達 94%,這明顯與減小ΔEST加快rISC來提高TADF效率的觀點不符。所以,我們不能像對待純有機體系那樣,只用ΔEST來判斷TADF效率的高低。于是,我們應用量化方法和TVCF速率理論詳細計算了配合物的ηTADF=[ηISC?ηrISC/(1-ηISC?ηrISC)]?ηVFR,ηISC=分別是S1→S0的輻射和無輻射速率常數,kTr和分別是 T1→S0的輻射和無輻射速率常數,和分別是S1?T1的系間竄越和反系間竄越速率常數。從分子57和58的計算結果我們發現,除了不同,57 的~106s-1要比分子 58 的~104s-1大兩個數量級,從而導致前者的TADF效率偏低。這表明在金屬有機配合物中,TADF 效率不僅與 ΔEST有關,更依賴于的大小。為了分析的來源,圖 7(b)給出了兩個配合物的重整能的分布圖,相對于配合物57,配合物58中的O原子橋連的四齒配體骨架限制了結構扭轉變化,大大減小了重整能,致使減小了兩個數量級。因此,我們可以通過引入O原子或者亞甲基構建六元環,以此增強體系剛性,保持較小的T1→S0無輻射速率,進而得到高效率的TADF分子。

表 1 計算出的配合物57和58的S0、S1和T1態之間的相互轉化速率(s-1)以及TADF發光量子效率(括號中是實驗結果)Tab.1 Calculated interconversion rates(s-1) among S0, S1, and T1 states, and TADF quantum efficiency for complexes 57 and 58[33].The available experimental results are listed in parentheses

圖 6 (a)不考慮 的磷光效率ηVP R =kr/(kr+()灰色三角)和考慮了的磷光效率=kr/(kr++()紅色圓圈)與實驗值 ηp(exp.)的對比;(b)配合物 44的激發態失活途徑;(c)3ES→S0過程的重整能;(d)3ES→S0過程對重整能貢獻前五的內坐標(鍵長、鍵角和二面角)[33],顏色按貢獻排序:紅色 > 綠色 > 藍色 > 黃色 > 粉色。Fig.6 (a)Comparison between calculated =kr/(kr+)(gray triangle) or =kr/(kr++) (red solid circle)and experimental ηp(exp.).( b)Schematic pathways of excited-state deactivations of complex 44.( c)The reorganization energies of 3ES→S0 processes.( d)The internal coordinates( bond length, bond angle, and dihedral angle) with the first five largest contributions to the reorganization energies[33].The colors are sorted by contributions: red > green> blue >yellow > pink.

圖 7 (a)分子結構;(b)配合物 57 和 58 的重整能量對內坐標的投影;(c)體系的 ηTADF、lg()、lg()和 ΔEST[33].Fig.7 (a)Chemical structures of investigated complexes(.b)Projections of the reorganization energies onto the internal coordi?nates for complexes 57 and 58.( c)ηTADF,lg(),lg(), and ΔEST of investigated complexes[33].
基于以上得到的結構-性能關系,我們設計了一 系 列 配 合 物 59~67,計 算 的ηTADF、lg()、lg()以 及 ΔEST見 圖 7(c)。 這 些 配 合 物 的TADF 效率被預測在94%~99%。這是因為通過調控取代基的給受電子能力,或者將四齒配體變成C^C^C^N和C^N^C^C,促進HOMO和LUMO軌道的分離,保持較小的 ΔEST。在該前提下,減小會進一步提高TADF效率。配合物67的TADF效率最高,甚至達到99%。該工作將對未來的理論和實驗相關工作提供有意義的指導作用。
眾所周知,在有機體系中,小的ΔEST是實現TADF的前提條件。根據量子化學理論,ΔEST主要與HOMO和LUMO之間的重疊積分成正比,HOMO和LUMO軌道之間的分離會導致小的ΔEST。因此,采用給體-受體(D-A)結構,形成具有電荷轉移(CT)特征的激發態是獲得TADF材料分子的最有效途徑[70-72]。以此,大量的具有較強的分子內CT、分子間CT以及空間/鍵共軛 CT 的TADF材料分子被合成和研究[73-78]。但是,小的軌道重疊不可避免地使躍遷偶極矩減小,相應地導致較小的輻射速率和受激發射截面(σem)[79-80],極大限制了TADF材料的應用。最近,Hatakeyama等提出了具有大振子強度的多重共振熱激活延遲熒光(MR-TADF)材料,其中缺電子原子和富電子原子相反地排列在剛性的多環芳香骨架上,通過多重共振效應使得HOMO和LUMO有效分離,進而產生TADF所需的小ΔEST[81]。但是,由于 MR-TADF材料通常具有較小的rISC速率和斯托克斯位移,這會導致自吸收和湮滅,不利于產生有機激光。因此,在TADF分子中同時實現大的、和σem是一個挑戰性難題[82]。這方面已經有一些很優秀的工作,例如,喬娟、彭謙等通過J型Frenkel激子與CT激子的耦合來同時得到快的和較小的ΔEST,在近紅外區域實現了高效的TADF[83]。馬於光、楊兵等提出具有局域-電荷轉移雜化激發態(HLCT)的熱激子機制,在確保有效rISC的前提下又有較快的輻射衰減[84],取得了很好的效果。
這里,我們提出了一種新策略:對于具有局域躍遷(LE)特征的S1和T1的分子,在保持S1的LE特性和大ΔEST的前提下,調節更高三線態(如T2)來調控S-T之間的轉化,實現局域發光態的TADF(LE-TADF)。基于這個策略,我們利用具有不同電負性的取代基(—NO2、—CH3和—NH2)來調節二氟化硼衍生物(圖8)的激發態,成功地通過LE/CT組分的比例調節了T2能級及其與S1之間的轉化,同時保持了LE特性的S1及其與T1之間的大能隙。圖8給出了各種取代后二氟化硼衍生物的激發態性質、態之間的自旋軌道耦合常數及激發態的衰減和相互轉化速率。由圖8可知,隨著取代基給電子能力從 S-NO2、S-CH3到 S-NH2增強,T2的電子躍遷性質從CT、HLCT到LE的變化,其能級正像我們期待的從低于S1到逐漸靠近S1最后高于S1,T2-T1的能級差隨之變大,這表明三者將會表現出不同的激發態轉化和發光行為。
圖8中數據顯示,相對于常見的碳氫體系的弱SOC(<1.0 cm-1),這三種體系的SOC都比較大,有利于單重態與三重態之間的轉化。我們進一步通過TVCF方法結合MOMAP 程序定量評估了各個過程的躍遷速率。對于S-NO2,S1→T2→T1的轉換速率很快,并且 T2→T1的比 T2→S1的大兩個數量級,這明顯有利于T1的生成。因此,實驗中S-NO2只發射磷光[85]。對于S-NH2,具有相當的輻射和無輻射速率常數,我們預測表現出熒光。令人興奮的是,對于 S-CH3,S1→S0的是傳統熒光的典型值,并且比普通TADF(~106s-1)大兩個數量級[86-87],S1?T2→T1間較快的相互轉換表現出動態平衡分布。計算結果表明TADF來自LE態。到目前為止,無論從能級角度還是從動力學角度均定量預測了LE態的TADF。我們合成了SCH3,并得到其光譜發射位置隨著溶劑極性增加變化很小,證明了S1態的躍遷性質與溶劑極性無關,沒有CT或HLCT的溶液極性依賴特征[34]。理論預測和實驗結果均證明,該化合物是一種很有前途的有機材料放大自發輻射材料。
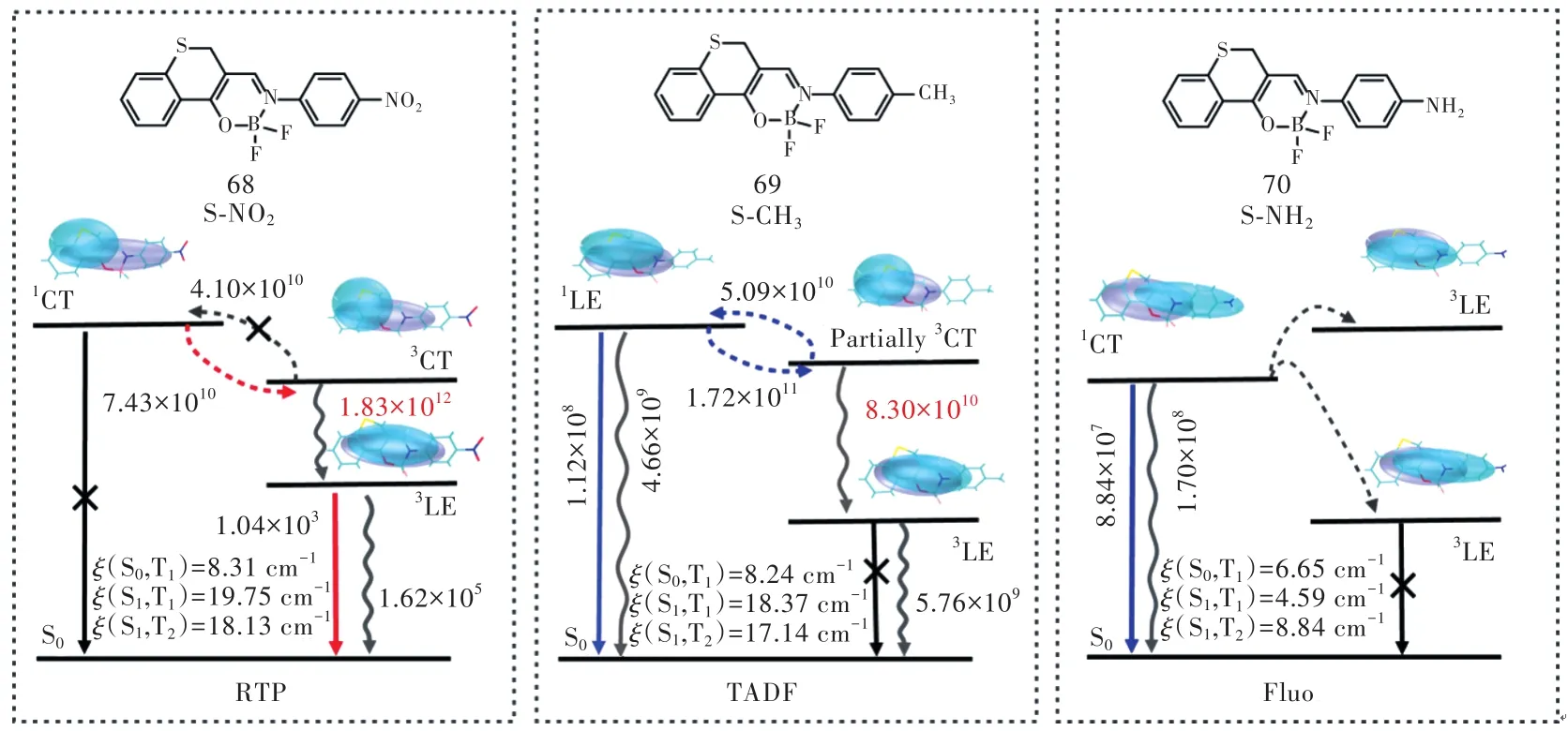
圖 8 體系的分子結構及其自然躍遷軌道分析、態之間的自旋軌道耦合常數(ξ)與激發態的衰減和相互轉化速率[34](各電子態的幾何平衡構型都是采用Gaussian 16程序包的B3LYP/6-31G*方法優化得到)。Fig.8 Chemical structures,natural transition orbital analysis,spin-orbit coupling constants between different states(ξ), rate constants of excited-state decay and conversion processes of investigated complexes[34] (The geometrical and electronic structures in all the involved electronic states were performed at B3LYP/6-31G* level in Gaussian 16).
基于以上結果,通過我們提出的分子設計策略,在二氟化硼衍生物中引入中性的甲基,保持S1的LE特征,通過調控T2性質與具有LE特性的S1之間的快速ISC/rISC過程,得到非D-A型結構的LE-TADF,這為高效率的TADF發展了提供了新的設計思路。
4 總結與展望
本綜述針對熒光、金屬配合物磷光、TADF材料,通過TVCF速率理論結合量化計算定量描述了有機體系的發光量子效率,揭示其發光理論,并提出高效發光的設計策略。我們提出的TVCF速率理論適合于大多數分子發光體系,但是不同的發光體系有著不同的發光機理。各類發光材料的結構設計著重于以下幾個方面:(1)對于熒光體系,我們根據分子內坐標對電子-振動耦合的貢獻,有針對性地改造、移除及重組分子片段來解開強烈的電子-振動耦合,從而減緩無輻射速率,增強熒光。( 2)對于藍光P(tⅡ)配合物體系,選擇合適的配體,避免3MC的生成;增強配體的剛性,避免大的結構弛豫,減慢無輻射躍遷。( 3)對于熱活化延遲熒光體系,含金屬的TADF分子,除了有較小的 ΔEST,必須增強體系剛性,保持小的,才能得到高效率的TADF。對于純有機TADF,通過分子設計,完全可以實現LE-TADF[88-89]。對于具有LE特征的S1和T1的分子,在保持S1的LE特性和大ΔEST的前提下,通過具有中等電負性的—CH3調節更高三線態(如T2)來調控S-T之間的轉化,從而在沒有典型D-A型結構的分子體系中實現LETADF。至此,我們的理論工作不僅能合理解釋諸多實驗現象,還能夠提出相應的分子設計策略,預測出高性能的OLED發光材料。
隨著有機發光領域的系列突破性進展,新現象、新概念、新機理和新材料不斷涌現,這給理論帶來了很大的機遇和挑戰。比如,如何實現激子利用率突破200%問題?如何理解有機共軛體系中的最低三重態高于單重態?如何設計三重態高于單重態的分子體系?如何解釋有機分子在聚集狀態下多重發射現象?如何設計和實現高效的深藍光體系?等等,這些問題尚待解決。我們一直在追求探索新模型、新理論和新方法的路上,期望可以為推動我國OLEDs產業的發展做出貢獻。
致 謝
感謝以下合作者的貢獻:王寓、肖小小、歐琪、林詩韻、張天、耿華、廖奕和帥志剛等。
猜你喜歡
科學大眾(2023年17期)2023-10-26 07:39:14
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
甘肅教育(2020年14期)2020-09-11 07:57:42
天天愛科學(2020年6期)2020-09-10 07:22:44
數學物理學報(2017年6期)2018-01-22 02:26:40
時代英語·高二(2015年1期)2015-03-16 00:08:11
中國衛生(2014年11期)2014-11-12 13:11:32
新高考·高一物理(2014年1期)2014-09-18 01:26:07
計算物理(2014年2期)2014-03-11 17:01:44
體育師友(2011年2期)2011-03-20 15:29:29
