基于熱活化敏化熒光的藍光材料與器件研究進展
2023-02-16 10:51:28黃天宇張東東
發光學報 2023年1期
關鍵詞:效率
王 琪, 黃天宇, 張東東, 段 煉
(清華大學化學系 有機光電子與分子工程教育部重點實驗室, 北京 100084)
1 引 言
有機發光二極管作為新一代顯示技術,具有自發光、寬色域、廣視角、對比度高、響應快等優點,被譽為21世紀“夢幻顯示技術”。經過數十年發展,OLED技術趨于成熟并成功產業化,被廣泛應用于手機、電腦、電視等各類顯示器。
OLED的研究核心在于開發高效穩定的材料和器件。第一代OLED[1-2]基于傳統熒光分子,具有穩定共價鍵、快速輻射躍遷速率(The rate ofradiative decay,kr)、低三線態(T1)能量,在電致發光器件中穩定性較好。但由于無法利用電致激發產生的三線態激子,器件內量子效率(Internal quantum efficiency, IQE)受到限制(≤25%),使得第一代OLED器件效率偏低。第二代OLED材料解決了三線態激子利用問題。磷光材料[3-4]引入Os、Ir、Pt等重金屬原子,利用重原子效應增大T1與S0之間的旋軌耦合常數(Spin-orbit coupling,SOC),使T1激子輻射躍遷回到基態過程變為自旋部分允許。器件中電致激發產生的單線態(S1)激子系間竄越生成T1激子隨后輻射躍遷發光,實現了100%的激子利用率。但磷光分子中配位鍵相對較弱且T1激子輻射躍遷緩慢,高亮度下三線態濃度顯著升高,加劇了三線態-三線態湮滅(Trip?let-triplet annihilation, TTA)及三線態-極化子湮滅(Triplet-polaron annihilation, TPA)等過程,進而損害器件穩定性[5]。目前,綠色和紅色磷光材料的效率及穩定性均已滿足實際應用需求,但藍色磷光材料由于激發態能量較高,穩定性仍然是瓶頸問題[6],OLED顯示產品中只能使用低效的藍色傳統熒光材料。因此,開發兼具高效率與長壽命的藍光材料成為OLED研究的“圣杯”[7]。
具有熱活化延遲熒光特性[8-9]的有機分子為解決上述問題提供了新思路。效率方面,TADF分子通過減小前線軌道重疊,降低單-三線態能量差(ΔEST)至0.2 eV以下,從而可利用環境熱使T態激子向S態激子反向系間竄越[10](Reverse intersys?tem crossing, RISC),由此實現100%的激子利用率。穩定性方面,TADF分子僅含有共價鍵,其鍵能大于磷光分子中的配位鍵;此外,TADF分子反向系間竄越速率(The rate of reverse intersystem crossing,kRISC)可以超過107s-1,比磷光分子的輻射躍遷速率快一個數量級以上,能更快速利用三線態激子,有效抑制三線態激子湮滅過程,減少高能中間體的生成。經過十余年發展,目前基于TADF材料的天藍光器件在1 000 cd/m2的亮度下效率超過20%,T97(器件亮度衰減至初始亮度97%的時間)達到110 h[11],取得了令人矚目的進展,展現了良好的產業化潛力。
但是,TADF材料多為電子給體-電子受體型電荷轉移態(Charge transfer, CT)分子,S1輻射躍遷較慢且發射光譜寬,限制了穩定性與色純度提升。為了克服上述矛盾,2012年,段煉等[12-13]提出了熱活化敏化熒光機制,如圖1(a) 所示。
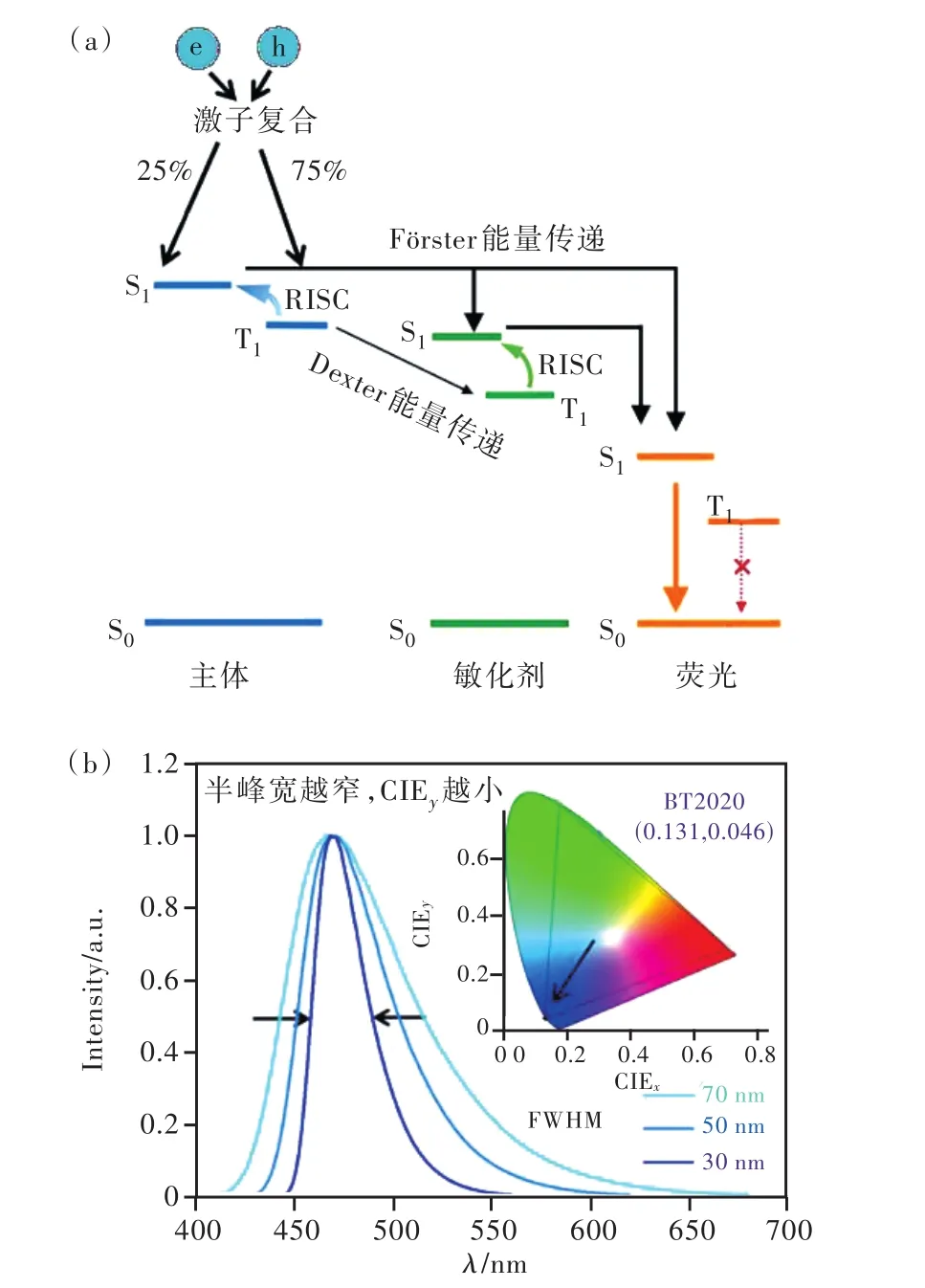
圖1 (a)TSF原理及激子能量傳遞路徑; (b)藍光光譜與CIEy的關系。Fig.1 (a)Principle and exciton energy transfer path of TSF.(b)Relationship between spectrum and CIEy
基于TSF機制的OLED器件選取TADF分子作為主體或敏化劑、窄光譜的硼氮分子或傳統熒光分子為染料,利用TADF分子快速的反向系間竄越速率實現三線態激子上轉換并通過F?rster能量傳遞(F?rster energy transfer, FRET)將激子能量轉移給染料,最終利用染料輻射躍遷發光。TSF機制由此實現發光層激子上轉換與輻射躍遷的功能分解,結合了TADF分子快速上轉換以及染料分子快速輻射躍遷優勢,激發態壽命顯著縮短,從而有利于提高器件壽命。TSF器件的另一個優勢是可選擇窄光譜熒光染料作為最終的發光材料。如圖1(b)所示,材料發射峰值相同,隨著光譜半峰寬(Full width at half-maximum, FWHM)變窄,器件光色可由天藍光藍移至深藍,TSF機制有望進一步提升器件壽命與色純度[14]。
近年來,隨著TADF敏化劑以及熒光染料的不斷進步,基于TSF的藍光器件效率和穩定性不斷提升[15]。本文圍繞穩定藍光TADF分子的設計開發綜述了近年來該類型分子的研究進展以及TSF藍光器件在壽命與效率方面取得的提升,并進一步指出未來發展的目標及面臨的挑戰。
2 高效穩定藍光TADF分子開發
TADF分子在TSF器件中具有激子上轉換功能,是實現三線態激子高效利用的關鍵,需要兼具較好的本征穩定性與快速的反向系間竄越速率。穩定性方面,由于藍光對應激發態能量較高,僅含有較穩定C—H、C—C、C—N化學鍵的氰基、1,3,5-三嗪、9H-咔唑等剛性基團及其衍生物被遴選出來,作為電子受體、電子給體應用于藍光敏化劑分子的構筑,衍生出三嗪-咔唑、苯腈-咔唑兩大體系。激子利用方面,由于藍光染料輻射躍遷速率普遍可以超過1×108s-1,遠大于TADF分子反向系間竄越速率[16],上轉換過程成為發光層激子利用的決速步。
加速激子利用、抑制三線態激子湮滅對提升器件穩定性至關重要。如圖2所示,在純有機TADF分子中,三線態激子上轉換至單線態受自旋禁阻限制為動力學緩慢過程,其壽命達到微秒量級。高亮度下長壽命三線態激子濃度上升,通過短程Dexter能量傳遞(Dexter energy transfer,DET)發生三線態-三線態湮滅、三線態-極化子湮滅等過程產生高能中間態,進而使材料化學鍵裂解、器件老化。因此研究者一方面引入惰性位阻基團屏蔽發光核心,減小相鄰分子間前線軌道重疊抑制DET;另一方面則增大TADF分子反向系間竄越速率,加快三線態激子上轉換動力學過程,降低三線態激子濃度,從根本上抑制激子湮滅過程,進而降低效率滾降(Roll-off),提高器件穩定性。但是,電子給體-受體型TADF分子在光色藍移與提高反向系間竄越速率之間存在矛盾:光色藍移要求降低給受體的給電子與吸電子能力,并需要一定程度上減小給受體平面之間的扭轉角抑制分子內電荷轉移;高效上轉換過程要求保持較強的分子內電荷轉移,增大TADF分子前線軌道分離程度保證較小的ΔEST。如何平衡乃至解決這個矛盾成為高效藍光TADF分子開發的關鍵科學問題。本文選取近年來具有代表性的藍光TADF分子,總結了在該科學問題上取得的進展以及藍光OLED器件在色純度、穩定性與效率等方面取得的突破。
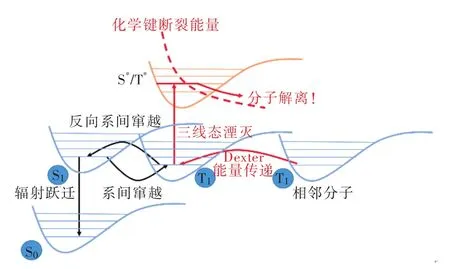
圖2 TADF分子的激發態失活過程示意圖。S0代表基態,S1/T1代表最低能量單線態/三線態,S*/T*代表高能單線態/三線態。Fig.2 Schematic diagram of excited state deactivation process of TADF molecule.S0 represents the ground state, S1/T1 represents the lowest energy singlet /triplet, and S*/T* represents the high energy singlet /triplet.
2.1 三嗪‐咔唑體系
根據費米黃金定則,分子kRISC可由以下公式進行計算[17-18]:
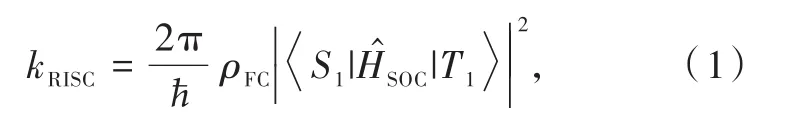
其中,||為 S1與 T1之間的 SOC 矩陣元;ρFC為弗蘭克-康登加權態密度(Franck-condon-weighted density),該項化簡后正比于 exp(-ΔEST/kBT),ΔEST表示單線態-三線態能量差,kB為玻爾茲曼常數。因此,相關分子設計策略圍繞減小ΔEST、增大SOC展開。
2011 年,Endo等[8]在Appl.Phys.Lett.發表了第一個應用于OLED的TADF材料PIC-TRZ。但該類型分子電子給體強度較大,難以實現光色藍移,后續藍光分子多以2,4,6-三苯基-1,3,5-三嗪為電子受體,電子給體鍵連在苯環上。圖3中結構式a所示的藍光TADF分子Cz-TRZ摻雜在DPEPO薄膜中,S1能級高達 3.18 eV,ΔEST接近 0.4 eV,器件最大外量子效率僅為4.5%。因此后續工作主要集中于在9H-咔唑的3,6位置連入取代基團(結構通式b)、對1,3,5-三嗪與9H-咔唑間橋連基團進行修飾(結構通式c)以及引入多給體(結構通式d),從而適當地使光色紅移并降低ΔEST,提高器件效率與穩定性。
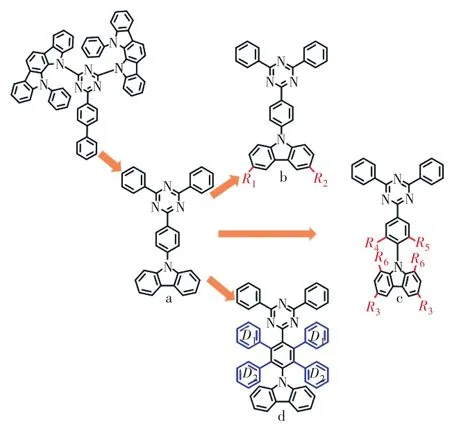
圖3 開發高效穩定三嗪-咔唑體系藍光TADF分子策略Fig.3 Development strategy of efficient and stable blue emission TADF molecules for Triazine-Carbazole sys?tem
2014年,Hirata等[19]以9H-咔唑、3,6-二苯基-9H-咔唑為取代基,設計合成了2c(R1= 9H-咔唑,R2=H)、2b(R1=R2= 9H-咔唑)、2a(R1=R2= 3,6-二苯基-9H-咔唑)一系列藍光TADF分子。如圖4所示,電子給體π共軛體系的延展導致了HOMO的離域化,增強了電子振動耦合并降低了前線軌道重疊程度,從而減小ΔEST,加快反向系間竄越過程。由于取代基接入2a等分子相較于Cz-TRZ光色稍有紅移,2b、2c甲苯溶液中發射峰為458 nm,2a為457 nm,但ΔEST顯著減小,其中2a僅為0.09 eV。以2a為染料、DPEPO為主體的器件取得了最優性能,器件色坐標CIE為(0.19, 0.35),為天藍光發射,最大外量子效率超過20%,1 000 cd/m2亮度下外量子效率仍接近11%,展現出較小的效率滾降。
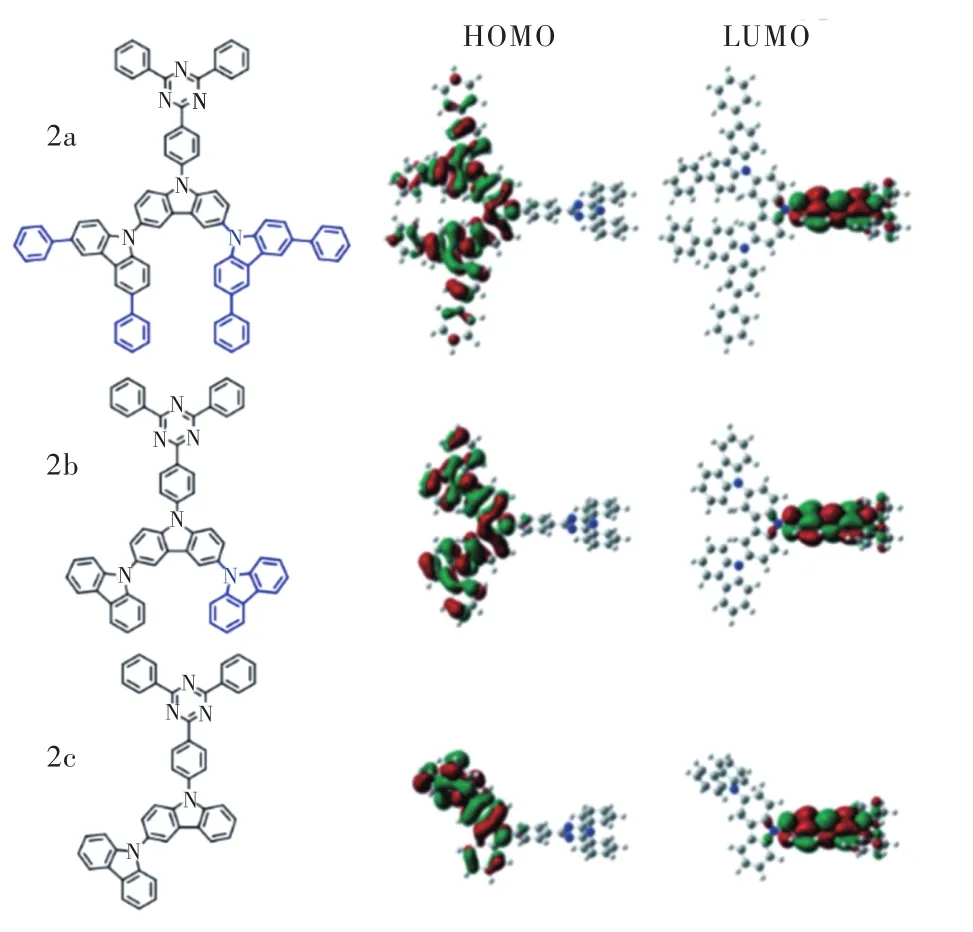
圖4 2a/2b/2c結構式與前線軌道分布Fig.4 Chemical structure of 2a/2b/2c and their frontier orbit?al distribution
2015年,Kim等[20]利用相同的策略分別在苯環上以間位連接的方式接入2個/4個9H-咔唑基團,設計了兩個穩定藍光TADF分子DCzTrz、DDCzTrz,通過促進HOMO離域化降低ΔEST。兩個分子ΔEST相近,約為0.25 eV,相較于Cz-TRZ大幅度降低。以DPEPO為主體,DCzTrz、DDCzTrz為染料的器件最大外量子效率分別為17.8%、18.9%,色坐標 CIE分別為(0.15, 0.16)、(0.16,0.22)。為提升器件穩定性,研究者進一步選用mCBP為主體,DCzTrz、DDCzTrz為染料,對比器件以光色相近的磷光分子為染料,發光層為mCBP∶Ir(dbi)3。500 cd/m2初始亮度下,DDCzTrz器件T80達到52 h,是對比器件的3倍,展現出三嗪-咔唑體系TADF分子良好的穩定性。
2017年,崔林松等[21]以甲基為位阻基團,設計合成了一系列具有TADF性質的深藍光分子,如圖 5(a)所示,其中R3=Me,R4=R5=R6=H 記為 Cz-TRZ1;R3=R6=Me,R4=R5=H 記為 Cz-TRZ2;R3=R4=Me,R5=R6=H 記 為 Cz-TRZ3;R3=R4=R5=Me,R6=H記為Cz-TRZ4。在9H-咔唑1,8位置引入甲基一定程度上增強了電子給體能力,Cz-TRZ3甲苯溶液中發射峰紅移至465 nm,而橋連基團修飾對光色影響較小,Cz-TRZ1、Cz-TRZ3、Cz-TRZ4甲苯溶液發射峰基本相同。位阻基團的引入增大了給受體平面的扭轉角,其中Cz-TRZ1二面角僅為49.8°,而Cz-TRZ2、Cz-TRZ3、Cz-TRZ4分別為86.7°、71.3°、82.3°。分子構型扭曲化促進HOMO-LUMO分離,降低ΔEST,加快反向系間竄越速率。未經修飾的為Cz-TRZ1為0.43 eV,相近光色的Cz-TRZ3、Cz-TRZ4分別為0.17 eV、0.15eV,反向系間竄越速率達到 1.53×104s-1與 2.83×104s-1,是 Cz-TRZ1 的 51倍與94倍。以Cz-TRZ3為染料、DPEPO為主體的器件取得了最優性能,器件最大外量子效率為19.2%,色坐標 CIE為(0.148, 0.098),接近標準深藍光發射。
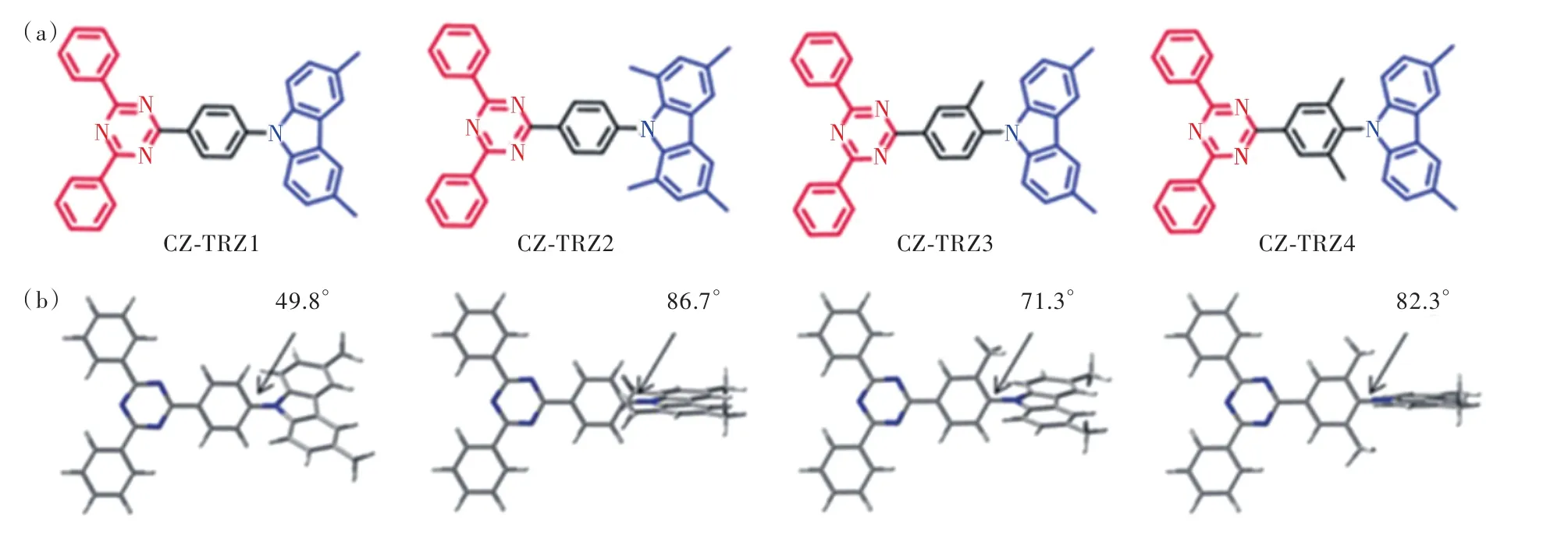
圖5 CZ-TRZ1~4的結構式(a)與優化構型(b)Fig.5 Chemical structures(a), optimized geometries(b) for molecules Cz-TRZ1-4.
但是,甲基化學活性較強不利于分子電化學穩定性,作為替代,更穩定且具有剛性結構的苯環被引入以調控分子的發光性質。2021年,Jeon等[22]選用苯環作為位阻基團設計合成了一系列TADF分子,其中R3=R4=R5=Ph,R6=H記為PPCz?Trz;R3=Ph,R4=R5=R6=H記為PCzTrz。苯環的引入同樣增大了給受體平面扭轉角從而提高了分子TADF性質,PPCzTrz分子ΔEST為0.16 eV,kRISC達到 8.02×104s-1;作為對比 ,PCzTrz分子 ΔEST為0.23 eV,kRISC僅為 1.44×104s-1。值得一提的是,該項工作以PPCzTrz、PCzTrz為敏化劑,v-DABNA為染料制備了OLED器件,發光機制以及能量轉移路徑如圖6所示。
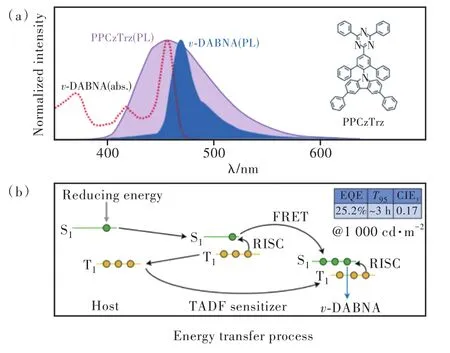
圖6 Jeon等工作的光物理特性(a)與發光機制(b),插圖為敏化劑PPCzTrz的結構式。Fig.6 Photophysical characteristics(a) and emission mecha?nisms(b) of Jeon et al.’s work.Inset shows the chemi?cal structure of the sensitizer PPCzTrz.
上述器件中主體三線態能量低于TADF敏化劑三線態能量、高于染料三線態能量。主體承擔激子傳輸與復合功能,TADF敏化劑完成三線態激子上轉換過程與三線態激子分配,部分三線態激子能量傳遞給主體再傳遞給染料,染料兼顧發光并輔助激子上轉換,由此保障器件高效率的同時降低主體三線態激子能量,提高器件穩定性。該機制激子利用關鍵步驟為TADF敏化劑承擔的三線態激子上轉換過程。以PPCzTrz為TADF敏化劑、v-DABNA為染料制備的器件性能最優,最大外量子效率超過33%,色坐標CIEy=0.17,1 000 cd/m2初始亮度下T95約 3 h,T50超過 150 h。
El-Sayed規則指出[23-24],為保持總角動量守恒,具有相同構型特征的S1和T1之間的ISC和RISC是自旋禁阻的。因此,若TADF分子S1和T1構型均為電荷轉移態(分別為1CT和3CT),即使降低ΔEST,kRISC提高仍有限,這是大多數D-A型TADF分子kRISC難以突破106s-1的原因。為解決這個問題,研究者在橋連基團上連入多重給體,一方面使HOMO離域化,減小ΔEST同時增強態混合,增大S1與T1之間的SOC;另一方面構建能級接近簡并的激發態,增加三線態激子上轉換通道。
2020年,崔林松等[25]利用該策略設計合成了一系列分子。當圖3通式d中Ds均為9H-咔唑時,記為5Cz-TRZ。該分子ΔEST僅為0.06 eV,兼具LE與 CT態的 T1使得 SOCS1-T1達到 0.4 cm-1,相較于常規TADF分子提升超過4倍。如圖7所示,T2-T1間能量差低至 0.24 eV,SOCS1-T2則超過 1 cm-1,使得T2-S1成為三線態激子反向系間竄越的有效通道。疊加以上因素,該分子kRISC達到1.5×107s-1。以5Cz-TRZ為染料、mCBP為主體構建的驗證器件,實現了486 nm的天藍光發射,器件EQEmax=29.3%,5 000 cd/m2亮度下 EQE=27%,效率滾降低于10%;壽命方面,高效快速的三線態激子上轉換抑制了TTA、TPA等過程,1 000 cd/m2初始亮度下器件T90接近600 h。另外,研究者以5Cz-TRZ為敏化劑、傳統熒光分子TBPe為染料制備了天藍光器件,得益于敏化劑快速上轉換速率,器件最大外量子效率超過20%且效率滾降較小。
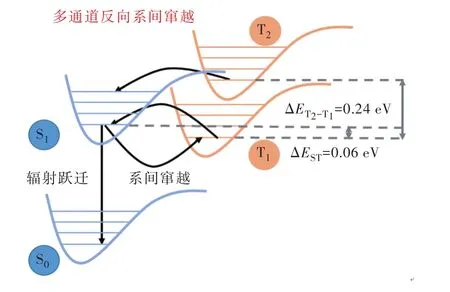
圖7 多通道反向系間竄越能級示意圖Fig.7 Schematic diagram of multi-channel reverse inter sys?tem crossing
2.2 苯腈‐咔唑體系
文獻中最早報道的苯腈-咔唑類藍光TADF分子為2CzPN[9],該分子在器件中發射峰約470 nm,最大外量子效率未超過10%。由于苯二腈作為電子受體過強,難以實現光色藍移,單苯腈-咔唑成為該體系中高效藍光代表性分子。由于圖8結構式a所示分子2CzBN-1[26]甲苯稀溶液發射峰過藍,ΔEST高達0.31 eV,且氮氣氛圍下PLQY僅有13%,后續優化工作主要集中于引入多重給體(結構通式b)、引入第二給體(結構通式c)、氰基對位鍵連受體(結構通式d),從而使光色適當紅移、降低ΔEST提升反向系間竄越速率、抑制非輻射躍遷提升PLQY等。
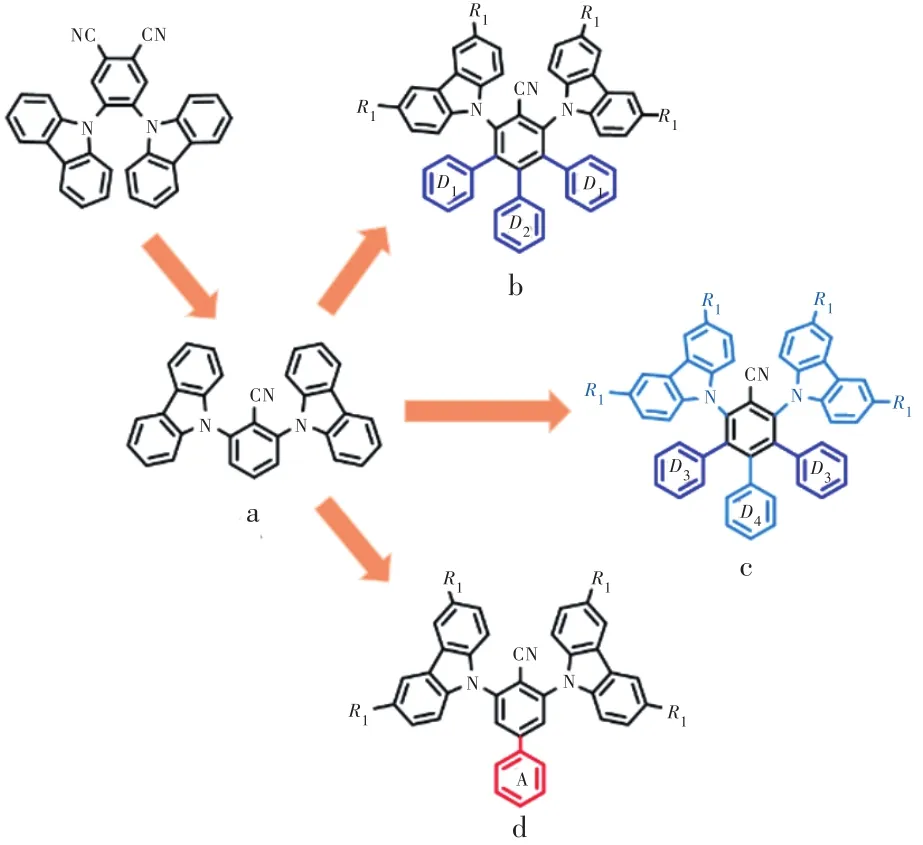
圖8 開發高效穩定苯腈-咔唑體系藍光TADF分子策略Fig.8 Development strategy of efficient and stable blue emission TADF molecules for Benzonitrile-Carbazole system
2016年,段煉課題組[27]率先報道了單苯腈咔唑類藍光材料,并引入多重給體,獲得了一系列從天藍光到深藍光TADF分子,圖8通式b中D1=9H-咔唑,R1=D2=H記為4CzBN;D1=3,6-二叔丁基-9H-咔唑,R1=叔丁基,D2=H記為4TCzBN;D1=D2=9H-咔唑,R1=H記為5CzBN;D1=D2=3,6-二叔丁基-9H-咔唑,R1=叔丁基記為5TCzBN(5TCzBN)。如圖9(a)所示,由于線性D-π-D結構有助于形成Cz+2自由基,該自由基與BN-相互作用形成離域激發態并增強1CT與3LE電子振動耦合,因此可以增大分子反向系間竄越速率[28]。
另外,基于B3LYP雜化密度泛函的計算表明,存在多個中間三線態陳列于整體分子S1-T1之間,這些中間三線態可能對激子上轉換有一定幫助。以 5CzBN 為例,如圖 9(b)所示,源自 m-3Cz?BN和4CzBN的中間三重態位于整體分子5CzBN的S1-T1之間,可能是5CzBN具有較大反向系間竄越速率的因素之一。2019年,Adachi 團隊[29]通過瞬態吸收證實了中間三線態的存在,它們來自于具有多重給受體分子的部分分子結構,為理論計算提供了實驗證據。結合以上因素,4CzBN、4TC?zBN、5CzBN、5TCzBN反向系間竄越速率均有一定提升,其中5CzBN達到1.13×105s-1,5TCzBN達到1.81×105s-1,且由于多重給體導致分子結構擁擠剛性增強,抑制了非輻射躍遷,5CzBN、5TCzBN分子的PLQY均超過70%。值得注意的是,位阻基團叔丁基的引入屏蔽發光核心,進一步抑制了三線態激子湮滅。如圖10(a)所示,5TCzBN分子HOMO分布被嚴格限制在咔唑上,并未分布到咔唑的3,6位取代基,這意味著叔丁基作為惰性位阻基團,不僅增大了分子間距離,也減小了相鄰分子前線軌道重疊,對抑制三線態激子湮滅過程起到重要作用。以5CzBN、5TCzBN為染料,mCBP為主體制備的驗證器件光色上兩者色坐標幾乎一致(CIEy≈0.40),都呈現天藍光發射;效率上5TCzBN器件EQEmax=21.2%,為5CzBN器件的1.26倍,證明引入叔丁基減少了電致激發下三線態激子的損失;而器件壽命對比更為明顯(如圖10 (b)),5TCzBN器件500 cd/m2初始亮度下T50達到770 h,是5Cz?BN器件的5倍。這是首個高效率、長壽命TADF天藍光材料,該策略也被廣泛應用于后續TADF分子設計。
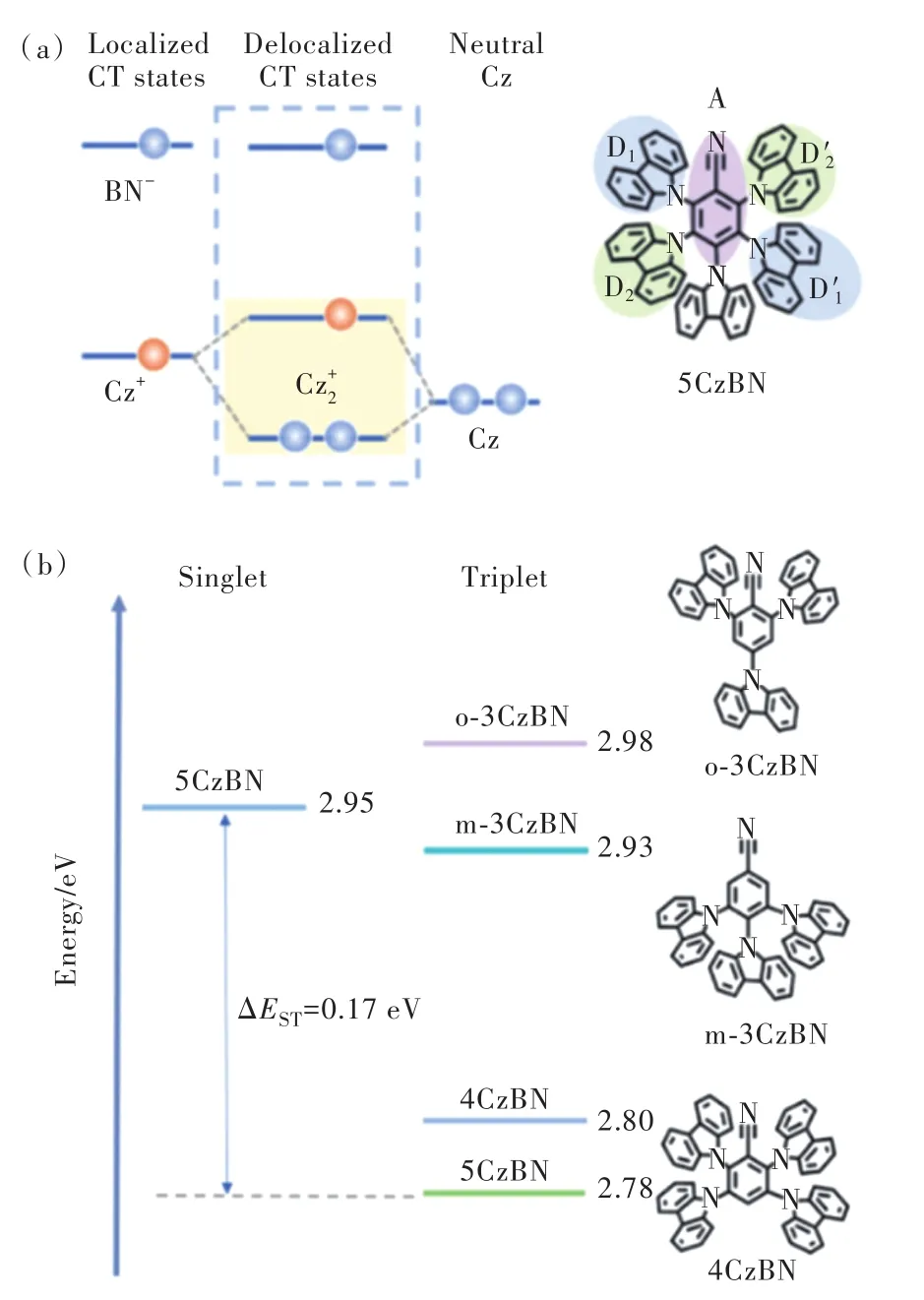
圖9 (a)5CzBN分子中離域CT態和線性D-π-D結構形成示意圖;(b)5CzBN衍生物的分子結構及能級圖。Fig.9 (a)Schematic illustration of the formation of delocal?ized CT states and the linearly positioned donors in the 5CzBN molecule.(b)Molecular structure of 5Cz?BN derivatives along with the energy diagrams.
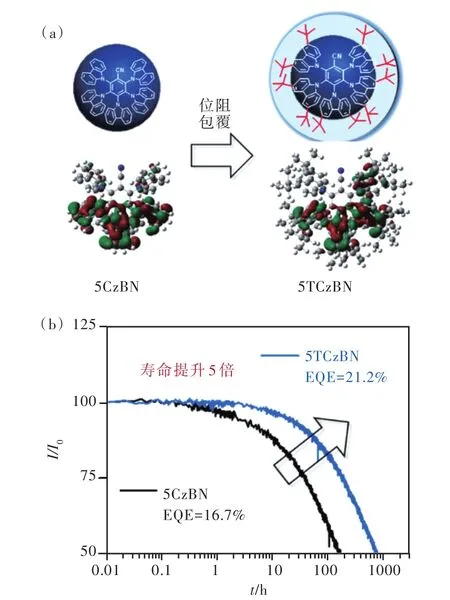
圖10 5CzBN及5TCzBN的化學結構及HOMO分布(a)、器件壽命對比(b)。Fig.10 Comparison of chemical structure and HOMO distri?bution(a) and device lifetime(b) of 5CzBN and 5TCzBN.
雖然D-π-D結構幫助增強了1CT與3LE電子振動耦合,但受限于3LE與3CT間較大的能量差,氰基咔唑體系進一步增大反向系間竄越速率仍有困難[30]。
2018年,Noda等[11]嘗試引入具有較低3LE能級的第二給體。如圖11所示,以5CzBN為原型分子,分別引入 3,6-二甲基-9H-咔唑(meCz)和 3,6-二苯基-9H-咔唑(DPhCz)作為第二給體設計合成了一系列藍光TADF材料,圖8結構通式c中D3=3,6-二甲基-9H-咔唑,D4=9H-咔唑,R1=H 記為3Cz2DMeCzBN;D3=3,6-二苯基-9H-咔唑,D4=9H-咔唑,R1=H記為3Cz2DPhCzBN;D3=9H-咔唑,D4=3,6-二苯基-9H-咔唑,R1=Ph記為2Cz3DPhCzBN。通過調整第二給體的數目和給電子能力,3LE態能級得到有效調節,顯著降低了3LE-3CT能量差。與5CzBN相比,所有具有第二受體的分子均顯示出PLQY的增加和TADF性質的改善。其中,3Cz2DPhCzBN分子3LE-3CT能量差為0.16 eV,相較5CzBN降低了0.16 eV,在ΔEST幾乎沒有變化的情況下該分子甲苯溶液中反向系間竄越速率達到 7.2×105s-1,超過 5CZBN 的 6 倍。以 3Cz2DPh?CzBN為染料的藍色OLED器件EQEmax=20.9%,色坐標CIE為(0.21, 0.44),呈天藍光發射,初始亮度 1 000 cd/m2下T97=110 h。
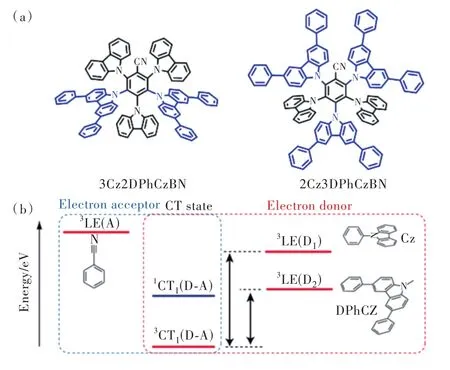
圖11 (a)3Cz2DPhCzBN及 2Cz3DPhCzBN分子結構式;(b)1CT1、3CT1和 3LE1能級排列示意圖。Fig.11 (a)Chemical structure of 3Cz2DPhCzBN and 2Cz3-DPhCzBN.(b)Schematic illustration of 1CT1, 3CT1,and 3LE1 energy level alignment for RISC.
2021年,Chan等[31]利用相同的策略設計合成了天藍光敏化劑HDT-1,分子結構為圖8結構通式 c中D3=3,6-二苯基-9H-咔唑,D4=間三聯苯,R1=H。該分子摻雜在mCBP薄膜中呈485 nm天藍光發射,PLQY為86%,反向系間竄越速率達到9.2×105s-1。以HDT-1為染料、mCBP為主體的器件最大外量子效率為 22%,CIE為(0.19, 0.40),初始亮度1 000 cd/m2下T95=28 h。為進一步提高器件色純度,以HDT-1為敏化劑、v-DABNA為染料構建了TSF器件。
圖12展示了典型的TSF器件能級排布與激子能量傳遞路徑。實現“天藍光敏化深藍光”的原因有兩點:一是具有CT性質的敏化劑激發態能量分布廣泛;二是v-DABNA分子小的斯托克斯位移使染料吸收光譜與敏化劑發射光譜間有較大重疊,建立了高效的FRET。較好的材料搭配降低了體系激發態能量,結合敏化劑、染料良好的本征穩定性與快速激子利用速率,驗證器件EQEmax≈27%、CIEy=0.20、1 000 cd/m2初 始 亮 度 下T95=11 h。
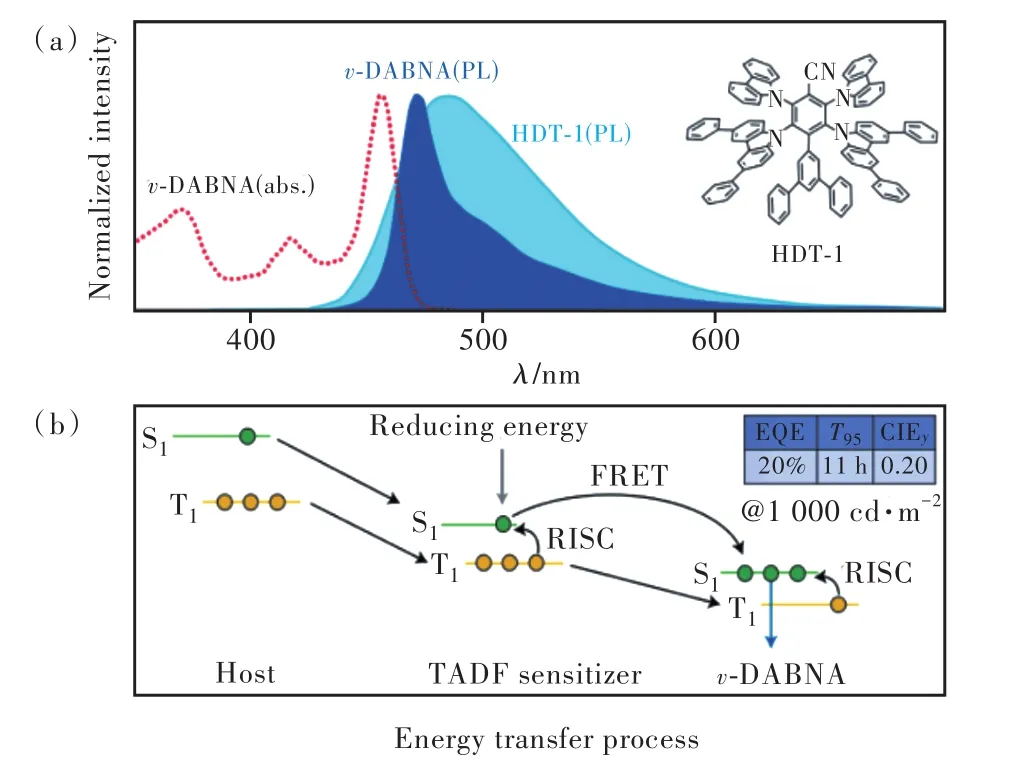
圖12 Chan等工作的光物理特性(a)與發光機制(b),插圖為敏化劑HDT-1的結構式。Fig.12 Photophysical characteristics(a) and emission mechanisms(b) of Chan et al.’s work.Inset shows the chemical structure of the sensitizer HDT-1.
然而,多重給體帶來的問題在于光色過度紅移,上轉換速率較快的分子幾乎都紅移至天藍光發射,色純度較差。如何保持高反向系間竄越速率的同時使光色藍移仍存在挑戰。
降低3LE與3CT間能量差的另一個策略為弱化電子給體強度,提升3CT能量。2020年,段煉課題組[32]提出使用苯腈代替氰基,弱化給體使光色藍移的同時,抬升1CT、3CT能量。由于3LE能量不受電子受體強弱影響,因此同樣起到了減小3LE-3CT能量差的作用。如圖13所示,p4TCz?PhBN的1CT能級與3LE能級相同,均為2.98 eV,而3CT也提升至2.88 eV,兩者間能量差僅為0.1eV。另外,p4TCzPhBN同樣具有線性 D-π-D、A-π-A結構,電致激發下形成的Cz2+自 由基 與 BN-相互作用形成離域激發態,增強1CT與3LE電子振動耦合,對增大kRISC亦有幫助。此外,值得注意的是,多重給受體引入使得p4TCzPhBN激發態能量接近簡并狀態,計算顯示p4TCzPhBN分子具有較小 ΔEST的同時,T2、T3能量位于 S1-T1之間,T2-S1、T3-S1可能成為輔助三線態激子反向系間竄越的有效通道。
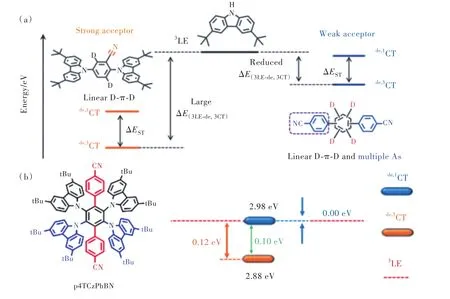
圖 13 (a)CzBNs和 CzPhBNs的相對能級圖;(b)p4TCzPhBN化學結構與該分子1CT1、3CT1和3LE1能級排列示意圖。Fig.13 (a)The relatively energy level diagram of CzBNs and CzPhBNs.(b)Chemical structure of p4TCzPhBN and the energy levels of p4TCzPhBN obtained from the spectra measured in toluene.
綜合以上因素,p4TCzPhBN分子kRISC超過2.3×106s-1,發射峰在甲苯稀溶液中藍移至456 nm。以該分子為染料、DPEPO為主體制備的驗證器件 EQEmax=22.8%,CIEy<0.2,在 1 000 cd/m2亮度下EQE仍超過20%,展現出極小的效率滾降。為進一步提升色純度,以mCPCz為主體、p4TCz?PhBN為敏化劑、t-DABNA為染料構建了TSF器件,最大外量子效率為32.5%,色坐標CIE為(0.13,0.12),100 cd/m2初始亮度下器件T80超過 3 000 h。
另外,多重給體導致分子量偏大,且分子構型擁擠,整體呈球形,一定程度上影響了光取出率。
2022年,段煉課題組[33]提出延展受體π共軛體系策略,設計合成了一系列深藍光TADF分子,圖8結構通式d中R1=Ph,A為苯環記為2PCzBN-Ph;R1=Ph,A為對叔丁基苯記為2PCzBN-tPh;R1=Ph,A為對氟苯記為2PCzBN-FPh。該策略通過減少咔唑受體數量增強分子平面性,提升了材料發光偶極水平取向,同時在氰基對位引入吸電子基團擴大受體共軛,增大LUMO離域程度,提升了材料TADF性質。LU?MO離域化一方面使前線軌道重疊減小,從而降低ΔEST,另一方面增強了1CT與3LE電子振動耦合,如圖14所示。據此設計的2PCzBN-FPh反向系間竄越速率達到8.31×105s-1,超過多重給體型分子5CzBN的7倍,且甲苯溶液中光色藍移至454 nm,分子水平取向達到84%,PLQY超過90%。
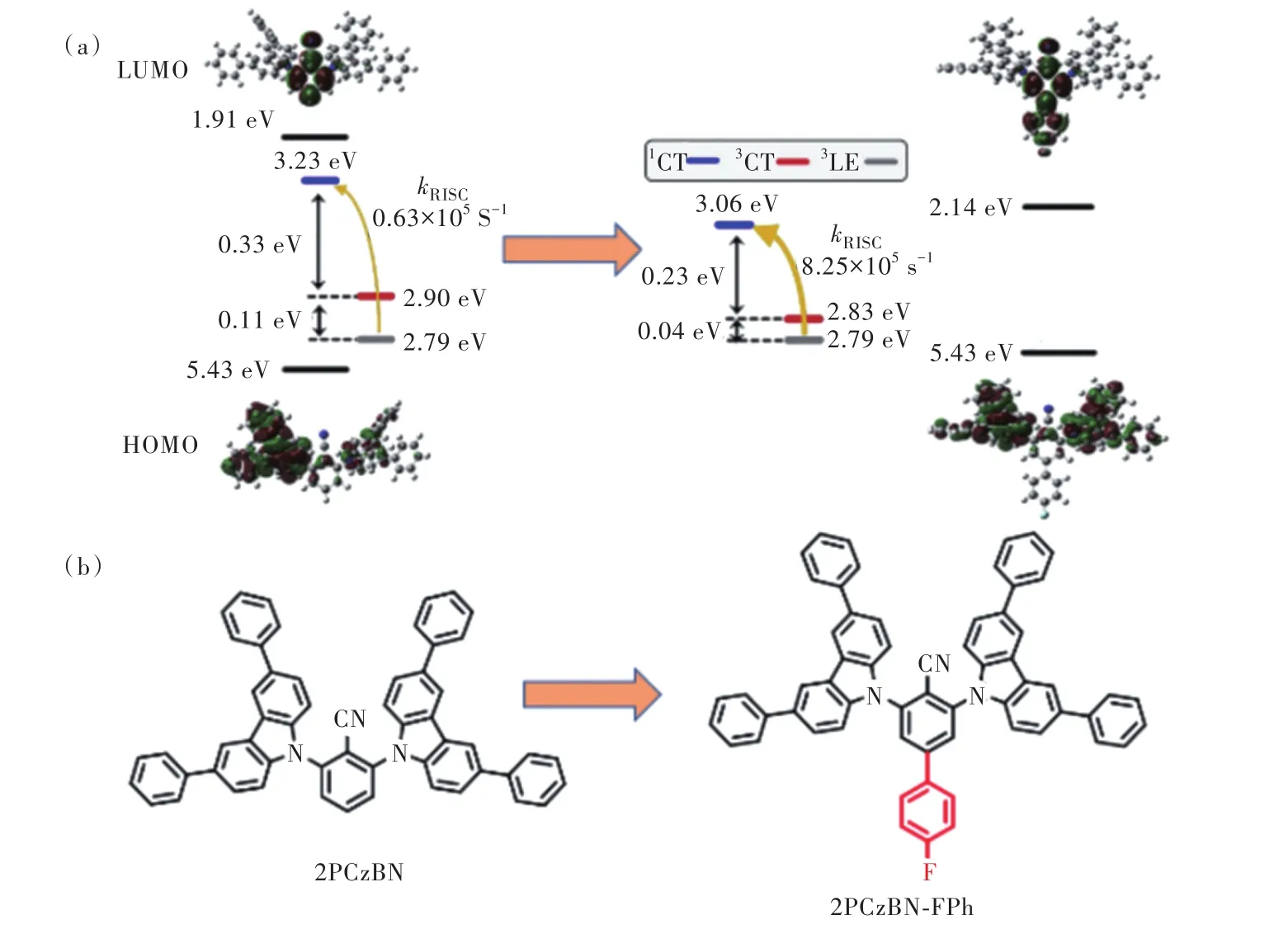
圖14 2PCzBN衍生物的前線軌道分布與計算能級排布(a)、化學結構(b)。Fig.14 The calculated distributions of their frontier molecular orbitals as well as the energy levels(a), and the structures(b) of the 2PCzBN derivatives.
以2PCzBN-FPh為染料摻雜在主體PPF中構建的驗證器件 EQEmax=35.7%,CIEy=0.25,1 000 cd/m2下器件 EQE≈25%,T50超過 100 h,展現出較小的效率滾降與較好的器件壽命,為氰基/咔唑體系開發提供了新的范例,實現了性能突破。
2.3 其他穩定藍光TADF分子
傳統D-A結構的TADF材料給體與受體之間的共軛效應會產生能量低的分子軌道,光色顯著紅移,不利于藍光材料開發。空間扭曲的D-A結構[34]以及非共軛的σ橋連的D-σ-A結構[35]是抑制共軛保持藍光的常用策略。然而,上述結構均難以兼顧藍光材料效率和穩定性。
2022年,張東東等[36]提出了D-Void-A的抑制D/A共軛效應新策略:由于分子軌道的形成需要給體和受體各自軌道電子云的空間重疊,而理論計算表明占噸酮3號碳原子上沒有HOMO以及HOMO-1軌道的分布,給體基團與C3鍵連時能有效抑制給體-受體間HOMO共軛,轉而與能級更深的受體HOMO-2形成分子軌道從而拉大發光帶隙(圖15)。相較于引入σ鍵打斷共軛,D-Void-A結構的分子具有一定的HOMO-LUMO重疊,振子強度更大且具有較強的分子骨架剛性,有效抑制了非輻射躍遷。基于該策略設計的33PCX光色藍移至440 nm,PLQY達到92%,反向系間竄越速率為4.3×105s-1。以33PCX為染料、PPF為主體的器件最大外量子效率為27.5%,CIEy=0.252。為進一步提升器件色純度,以33PCX為敏化劑、v-DABNA為染料構建了敏化器件,色坐標CIEy=0.19,100 cd/m2初始亮度下T95超過 650 h,證實了占噸酮單元作為TADF分子受體的良好穩定性,為后續工作提供了新的分子設計思路。
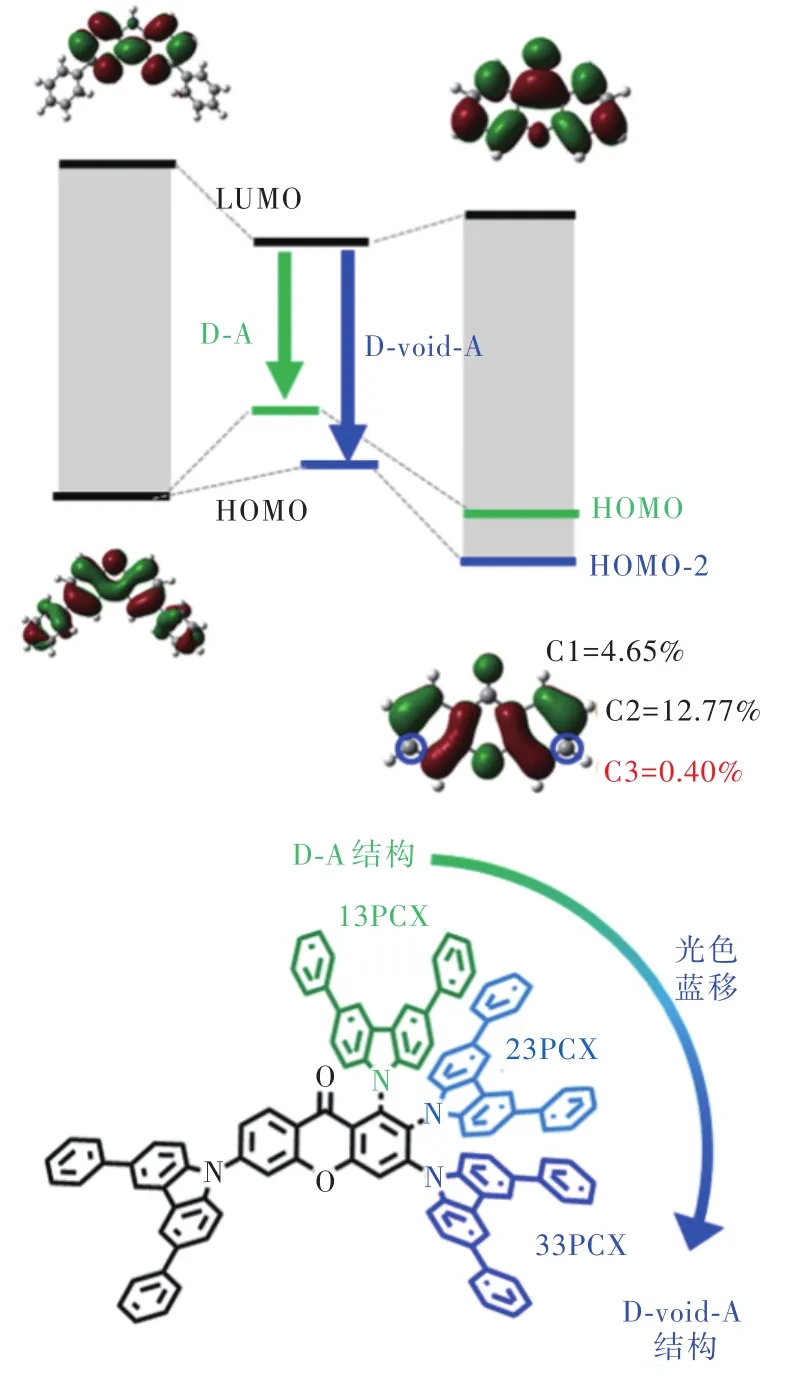
圖15 D-void-A結構示意圖及占噸酮-咔唑敏化劑分子Fig.15 Structure diagram of D-void-A and xanthone-carba?zole sensitizers
3 總結與展望
圍繞藍光敏化劑分子開發和TSF器件優化兩個核心課題,研究人員為構建高效穩定藍光OLED器件付出了諸多努力,在多個子課題上取得突破。提高材料本征穩定性方面,利用重水將TADF分子中C—H鍵氘代為C—D鍵,抑制因化學鍵振動造成的非輻射躍遷并淺化激發態勢能面,提高PLQY同時抑制分子處于高能激發態時的化學鍵裂解,從而提高敏化劑的穩定性[37];高效敏化劑開發方面,通過增強態混合、減小1CT-3CT-3LE能量差、構筑多重通道三線態激子上轉換、開發新給受體[38]、引入空間電荷轉移[39]等策略,打破TADF分子光色藍移與提高上轉換速率之間的制約,深藍光TADF敏化劑kRISC達到甚至超過106s-1,有效降低高電流密度下三線態激子濃度,抑制了激子湮滅過程;窄光譜染料設計方面,于稠環芳烴結構中引入硼(B)、氮(N)、氧(O)等多種或單一雜原子,降低前線軌道在化學鍵上的分布來抑制鍵長變化導致的振動耦合與構型弛豫,開發了新型高發光效率、窄光譜多重共振(Multiple resonance, MR)分子,提高了器件色純度;抑制發光層激子損失方面,引入剛性大位阻基團屏蔽發光核心,減小相鄰分子間前線軌道重疊,抑制Dexter能量傳遞,減少發光及能量傳遞過程中的激子損失;器件優化設計方面,合理設計發光層材料搭配優化器件能級排布,避免激子直接被染料捕獲、降低體系激子能量,提高器件效率的同時延長器件壽命。目前基于氘代敏化劑的深藍光TSF器件 CIEy~0.19,1 000 cd/m2亮度下T80超過 450 h,外量子效率達到16%,由于疊層器件、頂發光設計等器件工程能進一步優化發光色純度、提高器件效率、延長器件壽命[40],全氘代器件結合器件工程將助力藍光TSF OLED向產業化目標邁出一大步。
當然,TSF OLED產業化仍面臨巨大挑戰。為了滿足高分辨率4K/8K顯示器的廣色域標準,藍光OLED色坐標要求降低至(0.131, 0.046),這需要在目前工作的基礎上進一步藍移,開發發射峰小于470 nm、反向系間竄越速率大于106s-1的穩定敏化劑,光譜半峰寬小于18 nm、發射峰在460 nm左右的高效發光染料,以及與之適配的寬帶隙雙極性主體。從與現有工藝的匹配而言,二元而非三元TSF系統值得進一步研究。二元TSF系統主體兼具激子傳輸、復合與敏化功能,從而簡化器件制備工藝、減少發光層激子能量傳遞造成的能量損失,降低器件工作電壓。另外,作為競爭機制,近年來藍色磷光OLED器件與磷光輔助的熱活化敏化熒光(Phosphor-assisted TADF-sensitized fluorescence, TPSF)器件發展迅速,目前效能最好的藍色磷光OLED器件[41]CIEy~0.2,在1 000 cd/m2亮度下外量子效率超過23%,T95達到150 h;效能最好的藍色TPSF器件[42]CIEy~0.17,在1 000 cd/m2亮度下外量子效率近26%,T95達到73 h,成為藍光TSF OLED產業化的重要競爭對手。不過,TSF OLED在材料成本、元素豐度等方面具有磷光材料無法比擬的優勢,為藍色磷光OLED以及TPSF器件開發的穩定的主體及傳輸層材料亦可提高TSF OLED的性能。相信三者之間的競爭將促進高效、穩定的藍光OLED產業化。
猜你喜歡
瘋狂英語·初中天地(2021年5期)2021-07-21 02:24:28
甘肅教育(2020年14期)2020-09-11 07:57:42
中學生數理化(高中版.高考數學)(2020年5期)2020-06-02 09:19:08
商周刊(2017年9期)2017-08-22 02:57:49
遼寧經濟(2017年6期)2017-07-12 09:27:16
中國衛生(2016年9期)2016-11-12 13:27:54
時代英語·高二(2015年1期)2015-03-16 00:08:11
中國洗滌用品工業(2015年7期)2015-02-28 19:02:38
電子設計工程(2015年12期)2015-02-27 12:06:10
中國衛生(2014年11期)2014-11-12 13:11:32
