光電化學在有機合成中的應用
2023-02-11 02:15:58蔡浩然姚夏奕曹宇超陸澤鵬卞江
大學化學 2023年1期
蔡浩然,姚夏奕,曹宇超,陸澤鵬,卞江
北京大學化學與分子工程學院,北京 100871
1 引言
電化學有悠久的歷史,最早的電化學實例可以追溯到意大利科學家Volta,他在1800年發明了著名的伏打電堆[1]。在隨后兩百年的時間里,電化學領域吸引了一批又一批的有機化學家投身其中[2–7],到如今電化學已發展成為一個獨立的化學分支領域。電化學過程能夠生成常規手段難以得到的高活性中間體,如自由基、自由基離子和電子轉移復合體等。此外,使用電力也可以減少各種氧化還原試劑的使用[8],符合當前低碳減排的發展趨勢和需求。然而在電解過程中,由于在電極表面和溶液的界面處電子的有效傳遞效率較低,因此電化學生成的物種容易發生過度還原(氧化)、自由基偶聯以及電極鈍化過程[9]等不利的副反應。
與此同時,光化學在有機合成領域中也已取得了長足進展,在近二三十年里得到廣泛關注[10–15]。光化學和電化學類似,都通過外界輸入能量來活化底物[8],并且均以電子為中間物種[9]。然而,光化學亦有其局限性,如可見光能量太低,不能完成諸如由CO2和H2O制備葡萄糖等很多反應[16–18]等。因此,隨著光化學和電化學的興起,有機合成化學家們開始將光化學和電化學結合起來,使得二者之間可以取長補短(圖1)。
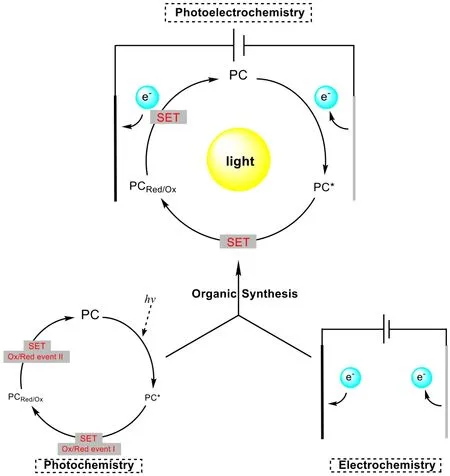
圖1 光電化學的示意圖
光化學與電化學相結合成為光電化學(photoelectrochemistry),它主要分為以下三種方法:電介光 催 化(electrochemically-mediated photoredox catalysis,EPC)、 去 耦 光 電 化 學(decoupled photoelectrochemistry,dPEC)以及界面光電化學(interfacial photoelectrochemistry,iPEC)。本文將著重介紹前兩種方法,具體原因會在第3.3節談及。
2 電介光催化(EPC)
2.1 EPC的概念
Moutet和Reverdy在20世紀70年代首次采用EPC方法,研究了由電化學方法產生的吩噻嗪自由基陽離子的光激發[19]。他們首先用電化學方法,將吩噻嗪(phenothiazine,PTZ)轉變為自由基陽離子PTZ?+然后通過光化學方法,使其與1,1-二苯基乙烯(1,1-Diphenylethylene,DPE)反應,得到產物1和2(圖2)。隨后,Rusling證明,4-氯聯苯的還原可以通過由光和電激發生成的蒽自由基陰離子形式的超級還原劑來完成[20]。
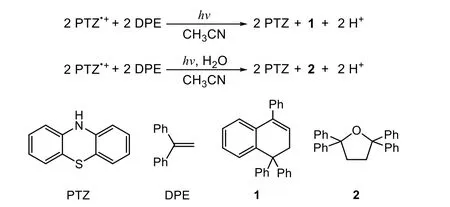
圖2 吩噻嗪自由基陽離子的光致激發
由此可見,電介光催化的基本思路就是:使用電流生成氧化(還原)性物種,然后通過光照對該物種進行活化,使其由基態成為激發態從而增強其氧化(或還原)能力。近年來,電介光催化的主要應用有[8]:i) 替代有催化劑毒性的化學氧化劑或還原劑;ii) 在溫和條件下生成高活性的反應中間體;iii) 活化具有高氧化還原電位的惰性化學鍵。
2.2 EPC的應用
2.2.1 C―C鍵的合成
C―H鍵能高,而外加電流可活化C―H鍵[21],這為C―C鍵的合成提供了一個思路。而使用光和電流共同作用,也能夠起到同樣的效果。下面介紹幾種不同的C―C鍵活化策略。
Ackermann小組報道了芳環的一種三氟甲基化方法[22](圖3)。他們選擇使用高氯酸(9-均三甲苯基-10-甲基吖啶) (9-Mes-10-methyl acridinium perchlorate,[Mes-Acr+]ClO4?)作為光催化劑,并以價格相對低廉的KOAc作為電解質,在室溫條件下,在芳環上成功地引入了三氟甲基。在這個例子中,電化學和光化學步驟以串聯的方式進行,使得催化劑量的染料催化劑活化,從而推動甲基化的反應順利進行。這正是EPC與dPEC之間的重要差別之一。
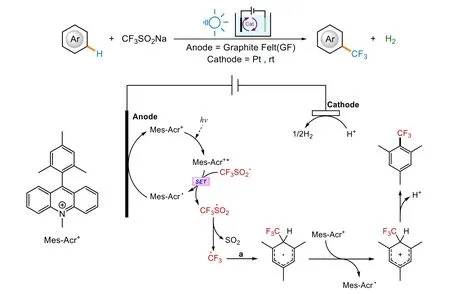
圖3 光電化學三氟甲基化及可能機理
三氟甲基自由基比較穩定。那么,有沒有什么辦法能夠通過生成一個相對來說不穩定的烷基自由基來實現C―C鍵的合成呢?廈門大學徐海超小組用羧酸解決了這個問題[23]。利用CO2作為氣體小分子的強可離去性帶來的反應熵增效應,推動烷基自由基的生成,可使烷基化反應得以順利進行。他們以取代喹啉3和環己酸4為原料,并以CeCl3?7H2O作為光催化劑、nBu4NCl為支持電解質,在392 nm的LED燈下反應17 h,最終以91%的產率分離出產物5 (圖4)。
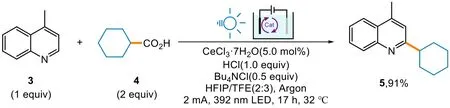
圖4 喹啉的烷基化
他們也給出了另一種烷基化方法[24],通過1,4-二氫吡啶衍生物以氧化芳構化的形式來提供烷基自由基。以藍光和2 mA電流激發以及在光敏染料Mes-Acr+的催化下,喹啉6和二氫吡啶物種7發生反應,能夠生成異丙基取代的產物8。
這類反應也應用了光敏催化劑的特性,機理大體類似。如他們亦發現酰胺9和叔丁基三氟硼酸鹽10可以在光敏化劑存在下制備吲哚衍生物11。該反應使用氧化的方式使得C―B鍵發生斷裂生成叔丁基自由基(圖5)。
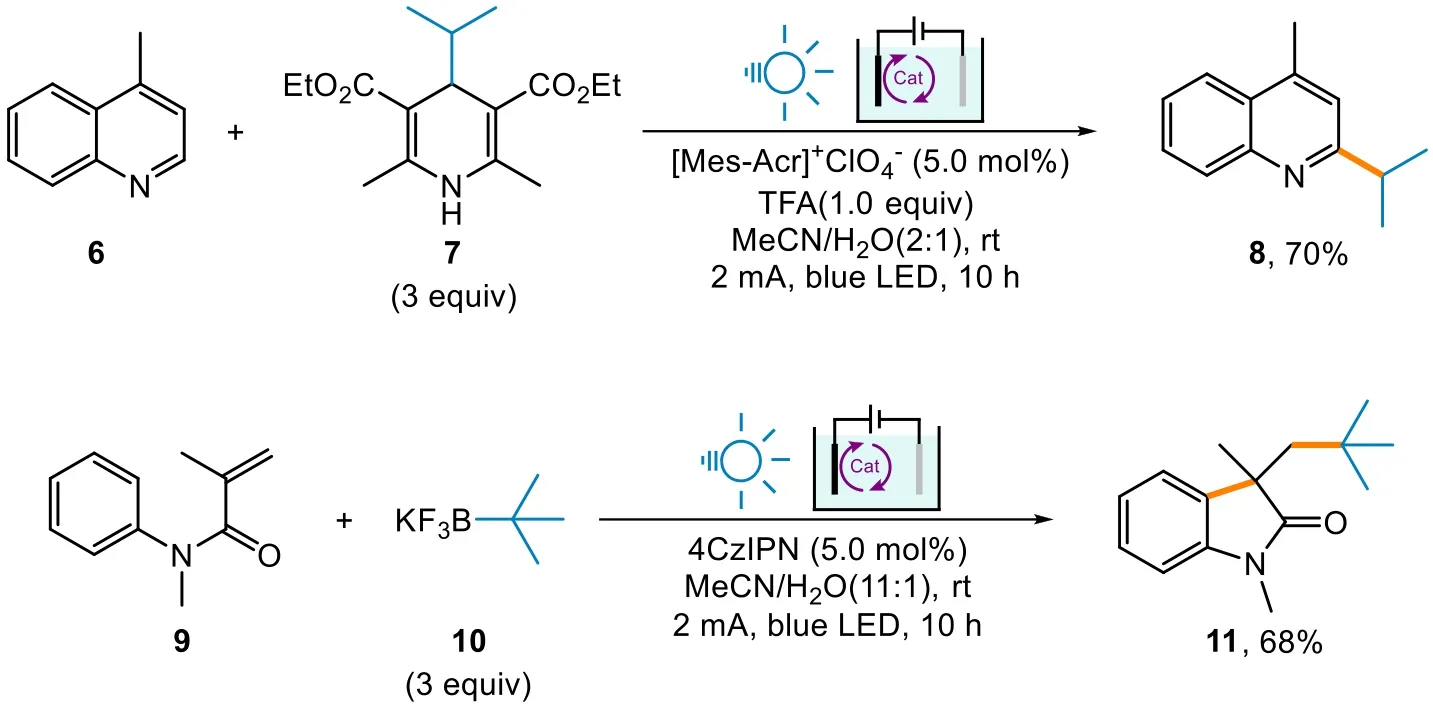
圖5 有烷基自由基生成的烷基化反應
2.2.2 C―N鍵的合成
目前已經有多種EPC反應的催化劑。如,Lambert小組使用三(2,6-二甲基環己胺基)環丙烷正離子12 (Trisaminocyclopropenium,TAC+)作為反應的光催化劑[25],成功活化了C―H和N―H鍵,從而合成了C―N鍵。他們對氧化電位很高的苯(Eox= 2.48 V)進行了研究。該反應在使用1.5 V電壓、23 W的熒光燈(compact fluorescent light,CFL)以及催化量的TAC+的條件下進行,成功實現了13的C―H和14的N―H之間的氧化偶聯反應,最終以65%的產率得到了產物15 (圖6)。
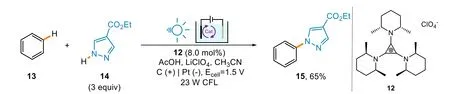
圖6 C―H鍵胺化
還有一類C―N鍵合成的例子是疊氮化反應。武漢大學雷愛文小組使用了EPC方法使疊氮化鈉參與反應,在光照和電流的作用下利用Mn化合物的助催化作用,成功地得到了疊氮化的產物17[26](圖7)。經過研究,他們提出了一種可能的機理。9-芴酮(9-fluorenone)經過光激發得到激發態中間體,該中間體與化合物16經過氫原子轉移(hydrogen atom transfer,HAT)過程后得到9-羥基芴自由基和芐位自由基,前者隨后在陽極上以單電子轉移(single electron transfer,SET)方式重新生成9-芴酮。MnII配合物與N3?離子配位,隨后在陽極被氧化,得到MnIII配合物。該配合物迅速同芐位自由基反應,生成疊氮基取代的產物,同時生成MnII配合物。
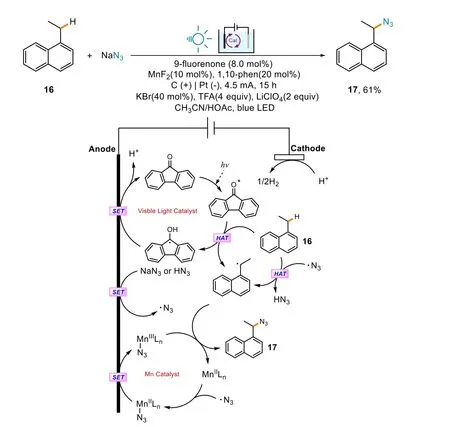
圖7 光電化學疊氮化與可能機理
2.2.3 氧化
EPC方法的本質是電子轉移,因此使用EPC方法也能夠進行氧化反應。林松小組提出了一種EPC氧化環己醇的新策略[27]:用光敏染料四乙酸核黃素(riboflavin tetraacetate,RFT)及硫脲18催化,環己醇29便能在電流和光的協同作用下以96%的產率(91%的轉化率)成功被氧化為環己酮20 (圖8A)。
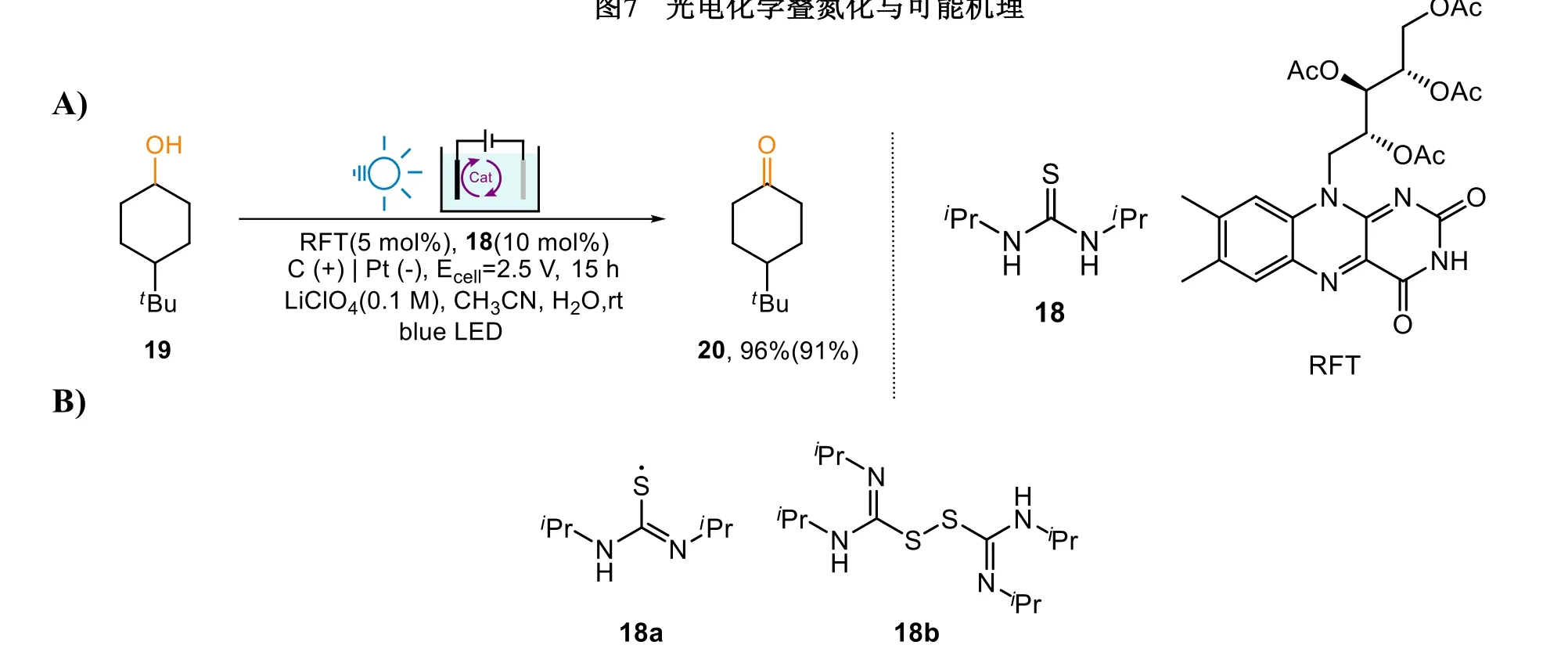
圖8 A) 光電化學方法對醇的氧化;B) 可能的中間體以及觀察到的偶聯副產物
硫脲18在這個反應中起到了關鍵作用。林松小組設計了多組實驗,發現RFT經光持續激發后可以持續產生巰基自由基18a,它會在后續步驟中充當氧化劑的作用。他們在反應中觀察到了18b中間體,表明有18a生成。
一般來說,一些高能中間體無法通過傳統方法生成,而EPC恰好能夠解決這個問題。實際上,單獨使用電流或者光照,或許已經可以解決這類問題;但是將兩者結合起來使用,則會有事半功倍的效果。
將光能引入體系往往需要特殊的光敏染料作為催化劑,這樣才能將光能轉化為活潑的化學能并傳遞給底物,從而使反應順利進行。此類染料的性質十分重要,但更為重要的是,這種光敏染料只需催化劑量就能夠支撐一個反應的進行,而不需要加入等當量的物料,因此制備成本大大降低了。
EPC方法近年來越來越為有機化學家所青睞。然而該方法仍有很多問題亟待解決:1) 為確定反應該使用什么波長的光需要反復的光譜分析得出結論,并且需要大量的實驗數據支持,程序極為繁瑣;2) EPC方法對反應設備、反應環境要求比較苛刻,因此要成為一種普遍的方法學還有很長的一段路要走。
3 去耦光電化學(dPEC)
前文討論過的EPC的特征是在反應過程中,電化學和光化學步驟分步串聯進行,來激活同一物種,最終生成目標中間體。而dPEC的策略則是電化學和光活化在互不干擾的兩步起作用,如此可以分別激活兩個反應機理中的不同組分。下面將介紹dPEC的一些應用。
3.1 C―C鍵的合成
dPEC最早可以追溯到Scheffold等人的工作,他們使用VB12物種來催化一個反直覺的α,β-不飽和羰基化合物的酰基化加成[28]。其中的一個例子使用乙酸酐來提供酰基,反應過程中成功實現了酰基自由基21的生成,這一關鍵步驟是通過光活化來實現的(圖9)。
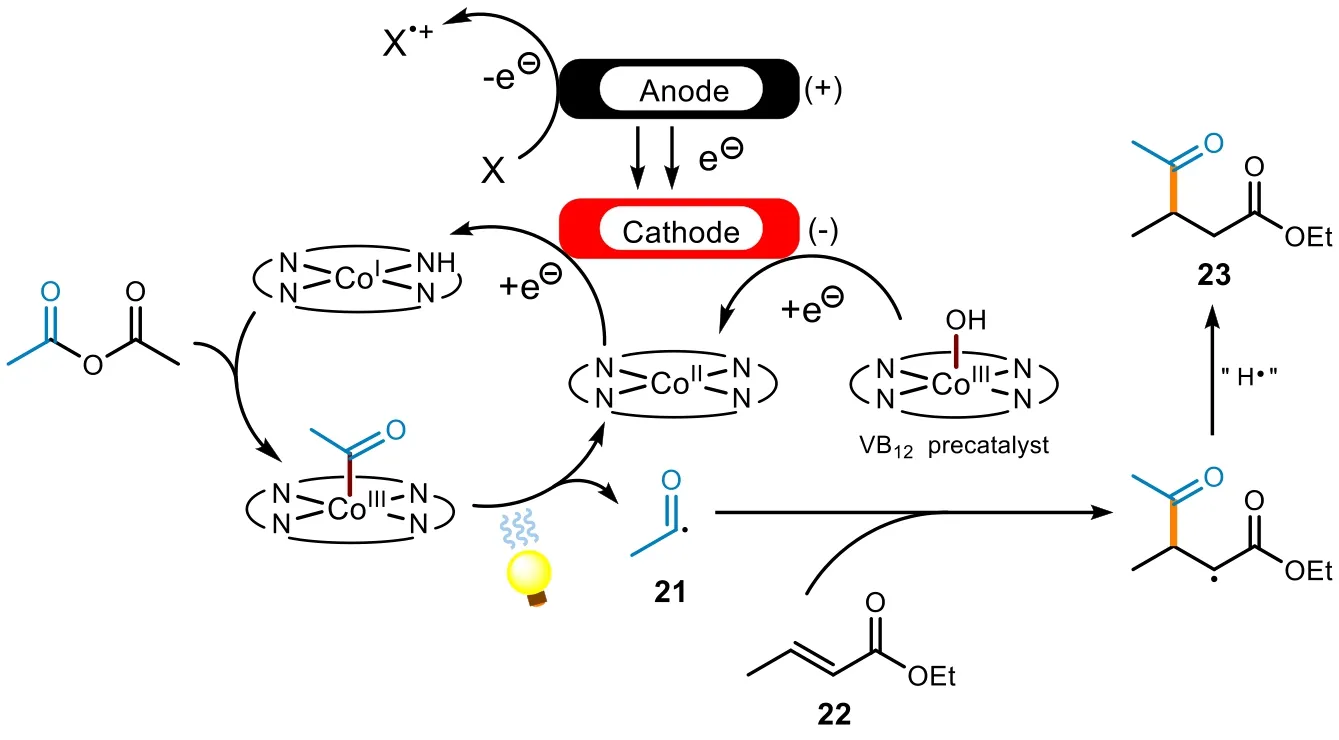
圖9 伴隨酰基自由基生成的酰基化反應機理
反應過程中,VB12物種在陰極得到兩個電子,生成CoI配合物。首先乙酸酐對該配合物進行氧化加成,生成酰基配合物物種,隨后在光催化下分解,生成CoII配合物與酰基自由基。然后,CoII配合物在陰極得到電子,還原成CoI配合物;而生成的酰基自由基21與α,β-不飽和酯22發生1,4-加成,生成的自由基中間體在電流推動下被氫原子自由基(H?)捕獲,最終便得到了極性反轉的Michael加成產物。該循環的關鍵一步是Co―C鍵的均裂,這一步是通過光的參與而進行的,而外加電流則起到啟動和維持反應的作用。
3.2 C―N鍵的合成
HLF (Hoffmann-L? ffler-Freytag)反應[29–31]在C―N鍵的合成中是常用的策略,該方法利用C―H鍵與N―H鍵之間的偶聯合成C―N鍵。這個方法最初只采用了電化學法。Stahl小組研究了如圖10所示的底物分子,總結了三種實現偶聯的策略[32],即:i) ET-PT-ET法(electron transfer-proton transferelectron transfer)來形成芐基正離子;ii) 堿促的質子耦聯電子轉移法(proton coupled electron transfer,PCET)來形成氮自由基;iii) 溴催化氧化法,以形成N―Br中間體。
Stahl小組認為這些方法都有其局限性。電化學方法無法避免對富電子的非底物物種的無效氧化,因此極有可能得到出乎意料的產物。為保證氧化的效率,很多小組均嘗試過新方法:使用化學計量的化學氧化劑,加入催化量的碘化物并加以可見光照射[33–36]。該法也能夠實現這一氧化過程,但仍然使用了外加的氧化劑。而Stahl小組則利用電化學代替了外加氧化劑。使用碘的好處是,它的氧化電位更低,因此它更加容易在較低的電壓下完成反應循環;與此同時,碘循環能和更多的底物兼容,具備很好的普適性,因此可以在更多種類的合成中發揮作用(圖10)。
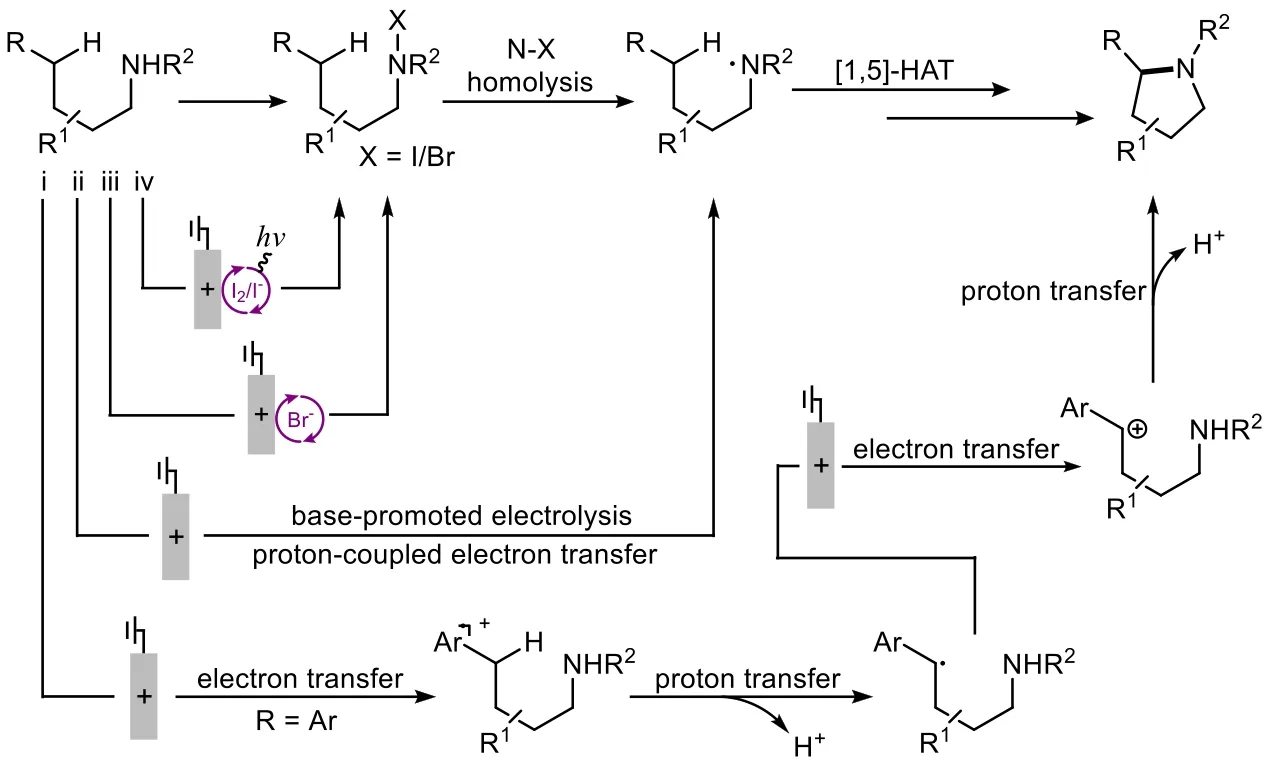
圖10 四種實現C―N鍵偶聯的方法
在合成化合物24b與25b的實驗(圖11)中[32],Stahl小組的方法體現出了其優勢。在該轉化過程中,24b分別在I、II、III條件下得到了理想的產率與轉化率,但生成25b的反應轉化率雖高產率卻不理想(如表1所示)。但是如果在圖11所示條件下使用TBAI催化該合成反應,25b的產率可達75%,分離產率可達72% (圖11)。
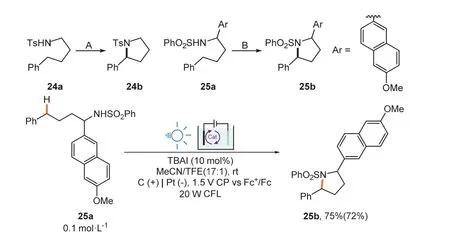
圖11 碘催化電氧化

表1 幾種C―H胺化電化學方法的比較
dPEC方法與EPC方法的本質區別在于光過程與電過程結合的緊密程度。顯然EPC方法將光過程與電過程作為連續步驟處理,實現了更為緊密的光電結合。
雖然dPEC本身并未將光、電的作用設計為緊密結合的連續步驟,但是光、電在催化循環中的作用都是不可或缺的。在dPEC方法中,通常光照在反應關鍵步驟起到作用,而電流則起到了啟動和維持反應進行的作用。電化學相較于一般的氧化還原犧牲劑有如下的優勢:1) 電化學可以調控氧化還原電位,不易出現過度氧化等現象;2) 電化學可以穩定氧化還原過程,并且持續時間更長,不會像化學氧化還原犧牲劑一樣隨時間推移而消耗,最終造成反應效率的下降。與EPC中光、電的協同作用相比,dPEC更傾向于以光化學作為反應主導,而以電化學為輔助。
在界面光電化學(iPEC)中光化學則更為邊緣化。iPEC方法旨在以光激發半導體電極來生成電流,從而催化反應的進行[37–39]。iPEC法的重點在于:1) 如何尋找合適的半導體電極,使其光電流剛好能夠催化反應進行;2) 什么反應恰好能夠在光電流的窗口電位內進行催化。本文側重于探討光電化學中光與電的充分結合對于反應過程的促進,而iPEC方法更關注電極材料的選擇,因此本文中不再對iPEC進行討論。
此外值得一提的是,對于光電化學領域的專業名詞,現在仍無通行的標準。在很多文獻中[8,40],EPC、dPEC、iPEC等名詞之間的分別并不是那么清晰。
4 光敏染料分子
相當數目的光電化學相關反應中使用了諸如Mes-Acr+或RFT等有機染料分子,因此找到合適的有機染料分子是PEC必須解決的問題。顯然,大部分染料分子的吸收峰都在可見光區,這使得它們極易形成分子激發態。但在催化循環中,只具備該性質并不足以成為PEC反應的催化劑。活化后的有機染料分子同時要具備合適的電位,才能氧化或還原穩定的C―H鍵或者N―H鍵。
本節針對現有PEC反應使用過的光催化劑進行了整理,并且對各種催化劑的電化學和光化學性質進行了統計分析。
4.1 兩種光敏分子催化的機理
K?nig小組對EPC方法提出了“SOMO-HOMO反轉”(SOMO-HOMO inversion)的概念[40]。無論是用于氧化還是還原反應,光電催化劑都需要經歷一個電激發過程與一個光激發過程以產生一個電介光氧化還原催化劑(electromediated photoredox catalyst)。例如,在電流作用下,還原劑分子的LUMO軌道上填入一個電子,該LUMO軌道變成SOMO軌道;隨后在光的作用下,HOMO軌道上的一個電子被激發到SOMO軌道上,最終表現出來的結果就像是HOMO軌道和SOMO軌道發生了“反轉”。例如9,10-二氰基蒽(9,10-Dicyanoanthracene,DCA)用作催化還原時與PTZ用作催化氧化時便是如此(圖12)。
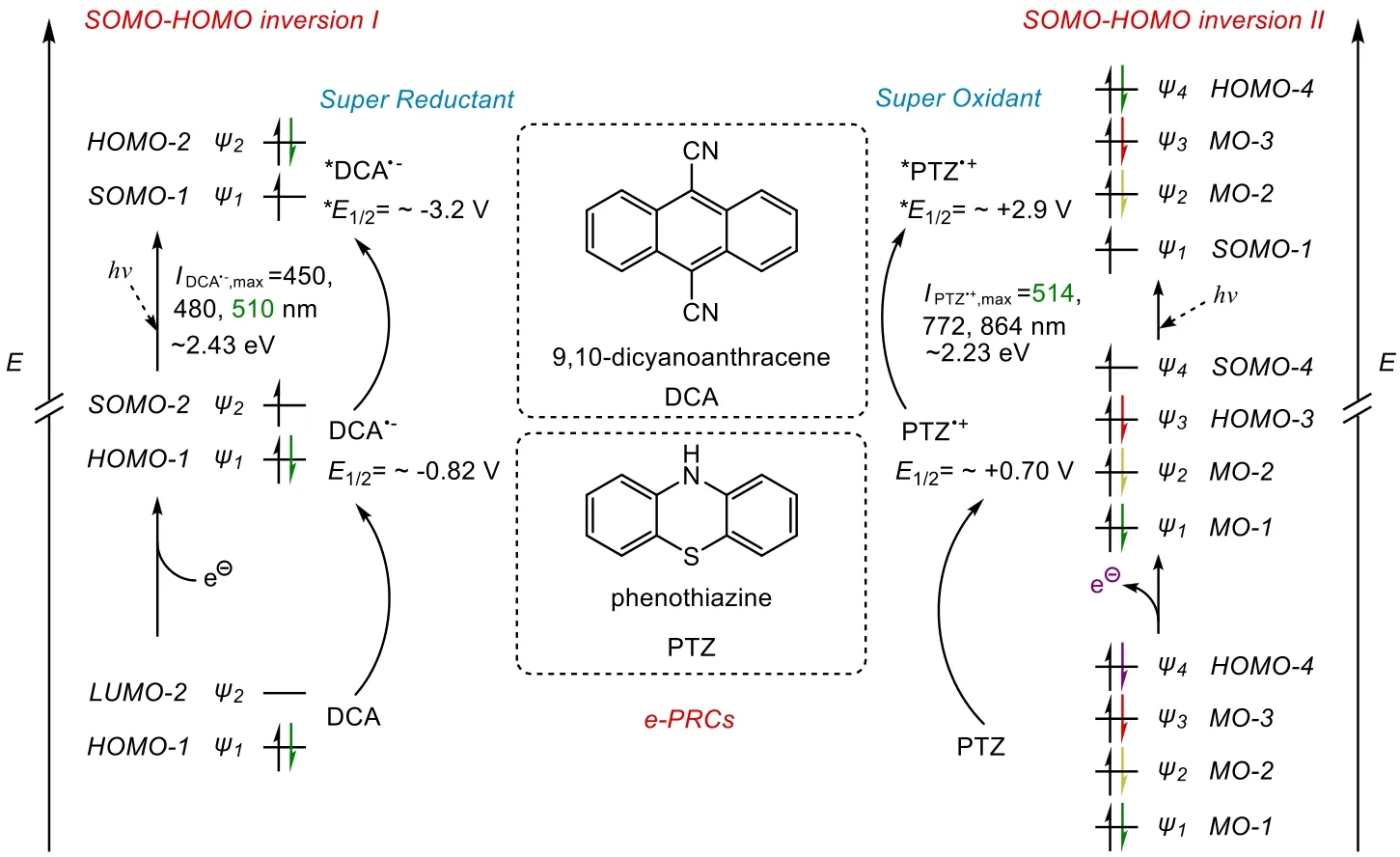
圖12 “SOMO-HOMO反轉”示意圖
除了以HOMO-SOMO反轉的方式生成超級氧化劑或還原劑,更多數的分子僅通過光激發的方式生成氧化還原劑,此時電流起到再生催化劑的作用。例如前文中提到的Mes-Acr+催化劑,就是以這種方式活化的。
4.2 幾種染料分子的光譜吸收峰和氧化還原電位
表2中匯總了多個染料分子(圖13)的吸收光譜數據與激發后的氧化(還原)電位,并作了統計。
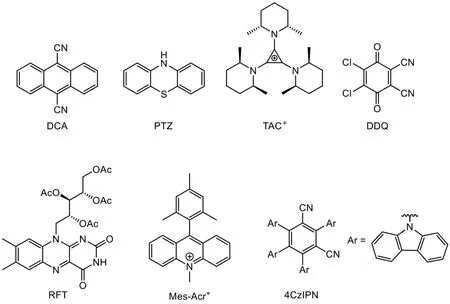
圖13 幾種染料分子的結構式
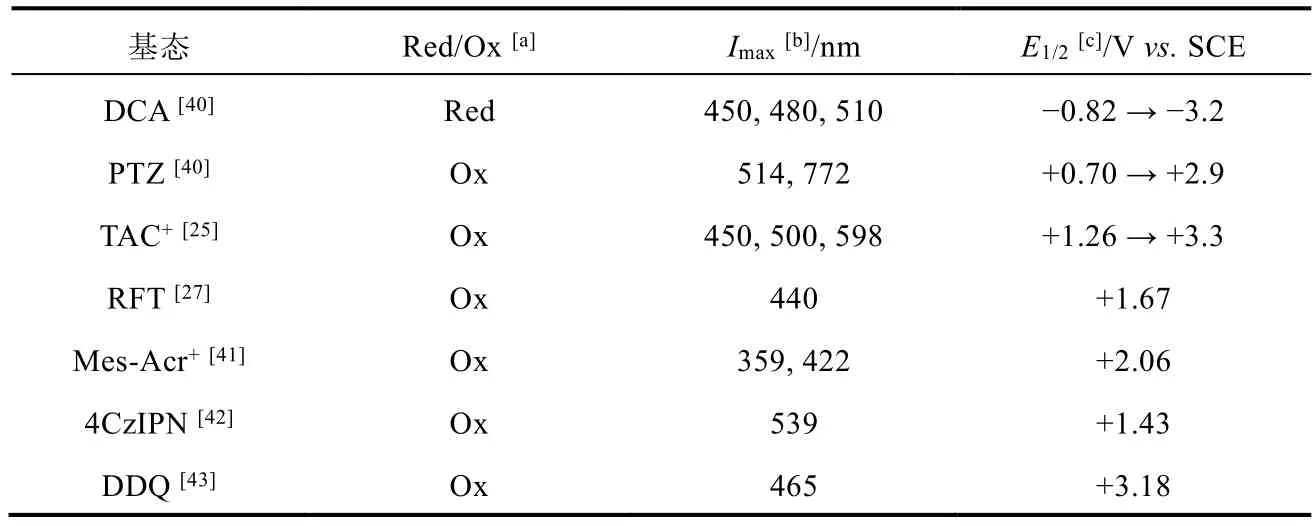
表2 染料分子的吸收峰與氧化(還原)電位
可以發現,電介光氧化還原催化劑相比光催化劑具備更高的氧化還原電位,這使得諸如2.2.3節中的脂肪醇氧化反應等需要高電位的氧化反應,以及諸如鹵代芳烴還原反應等需要強負電性的還原反應有了可能性。
這種高電位的產生和共軛體系的電子密度密切相關。例如,在DCA中,兩個氰基取代之后的蒽環骨架,由于氰基的吸電子效應,使蒽環具有一定的氧化性;而通過光、電的激發,可使其氧化性大大增強。這與基態2,3-二氯-5,6-二氰基苯醌(2,3-dicyano-5,6-dichlorobenzoquinone,DDQ)被激發為三線態3DDQ*[43]有相似之處。同時,在PTZ中,由于氮和硫為給電子基團,使芳環具有還原性;在光電激發后,PTZ便獲得了更強的還原性。
TAC+與Mes-Acr+同為陽離子,卻并未采取同一種催化機制。實驗證明,Mes-Acr+與TAC+在反應中所扮演的角色并不相同,但Mes-Acr?與TAC+卻比較相近。
據我們推測,這與電介光氧化還原催化劑*TAC2+·中的六個甲基的位阻效應相關。六個甲基的存在使得*TAC2+·的穩定性大幅增加,這是由于六個甲基的排斥使得還原劑無法接近*TAC2+·,因此后者壽命延長,生成電解光氧化還原催化劑。此外,TAC+本身的共軛體系不大且分布較為離散,其可能具備高的HOMO-LUMO能級差,因此采取直接的光激發機制并不可行。
Mes-Acr+則具備不同的結構性質。DFT計算表明[44],Mes-Acr+的均三甲苯基與吖啶基在空間中幾乎處于正交關系,其HOMO與LUMO分別主要分布于均三甲苯基與吖啶基上(圖14A)。若采取第一種活化機制,則均三甲苯基將發生電子轉移,隨后產生兩正電環正交相連的、高度不穩定的結構。因此直接的光致激發機制對于Mes-Acr+來說是更為可能的路徑(圖14B)。
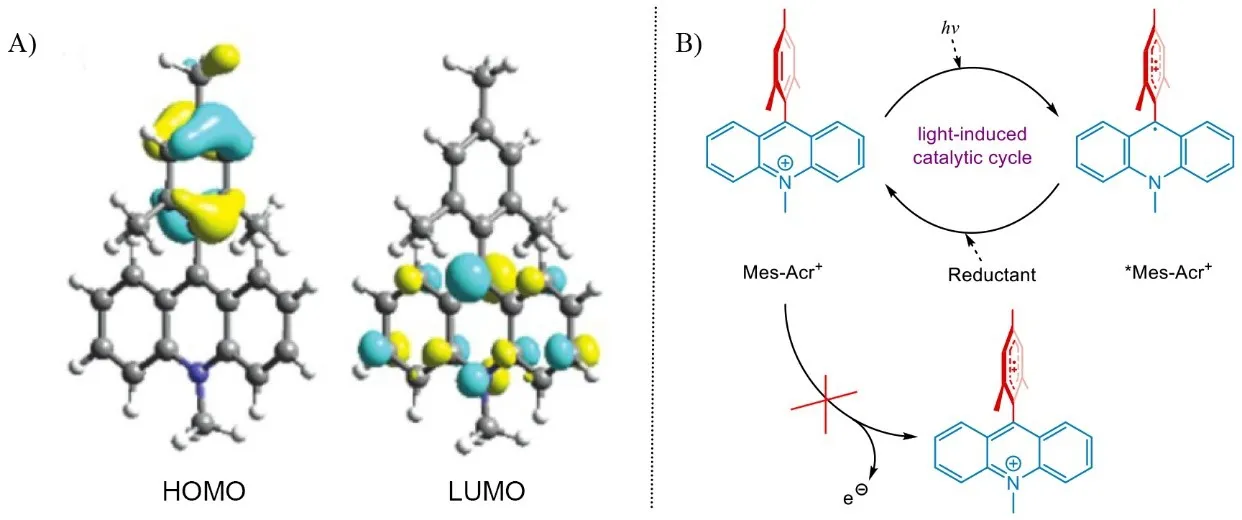
圖14 A) 通過DFT理論計算得到的Mes-Acr+的HOMO和LUMO;B) Mes-Acr+的光促催化循環
綜上,尋找能夠生成電介光氧化還原催化劑的光電催化劑應當滿足以下幾點要求:1) 具備大的共軛體系使得體系具備“SOMO-HOMO反轉”的可能;2) 依據電位要求對骨架進行修飾,使其具備順應電位需求的電子密度;3) 通過電化學方法完成電子轉移(electron transfer,ET)過程后在可見光區有吸收峰;4) 生成的電介光氧化還原催化劑具有一定的穩定性,能夠在反應體系中存在足夠長的時間以催化反應進行。相比之下,選擇采取第二種催化機理的光敏染料的標準并非十分苛刻,只需在光敏染料的基礎上尋找具有合適的氧化還原電位的分子即可。
5 結語
光和電對化學反應的作用殊途同歸,二者都是利用外界能量來對體系進行激發和活化,使反應得以跨越熱力學或動力學上的能壘,得以大規模、高選擇性、高環境友好性地合成產物。
從當今的化學學科發展來看,技術交叉和融合是整體的大趨勢。吸納和利用不同領域的知識、靈活運用各種方法有助于解決新出現的問題。在過去二三十年里,隨著傳統的有機化學方法已得到充分發展,而同時新的需求和挑戰不斷涌現,發展非傳統或非常規的有機合成方法已經成為未來一段時期內的重要發展方向。
就目前而言,光電化學的基本優勢[40]在于:1) 拓寬了單電子轉移(SET)反應的電位窗口限制;2) 采用更溫和的反應條件,具備更高的官能團耐受性與更高的反應選擇性;3) 提高了反應的能量利用率和原子經濟性。
目前,光電化學仍有很多局限。它的操作復雜、裝置稀缺且普適性低,以及具體到某個分子體系時,光電化學反應的位點與作用效果仍難以預見。正因如此,光電化學技術仍有若干重要技術問題有待解決。不過可以相信,在未來一二十年,光電化學將迎來一波快速發展期,并帶給我們更好的方法和對合成更好的理解。
猜你喜歡
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
科技知識動漫(2017年5期)2017-05-11 21:34:16
科技知識動漫(2017年4期)2017-04-15 22:24:55
科技知識動漫(2017年2期)2017-02-06 20:59:46
科技知識動漫(2016年10期)2016-10-18 20:35:00
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
Coco薇(2016年2期)2016-03-22 02:42:52
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
Coco薇(2015年1期)2015-08-13 02:47:34
