煤熱解過程中噻吩類硫化物遷移轉化機理研究進展
2023-01-07 00:47:42楊雙維夏源谷馬善為
煤炭學報 2022年11期
劉 吉,楊雙維,趙 微,胡 斌,夏源谷,馬善為,陸 強
(1.華北電力大學 新能源發電國家工程研究中心,北京 102206;2.華北電力大學 蘇州研究中心,江蘇 蘇州 215000)
煤炭是一種重要的化石燃料,其清潔高效利用對雙碳目標的實現意義重大[1-3]。熱解是煤炭重要的利用技術之一[4],也是燃燒過程的初始階段[5-6]。熱解過程中,煤在高溫環境下發生一系列物理和化學變化,最終得到煤氣、焦油和焦炭產物[7]。硫元素是煤炭中一種重要的雜質元素[8-10],在煤熱解過程中會發生復雜的遷移轉化從而進入固液氣三相產物中[11],遷移至焦油和焦炭中的硫元素將會導致焦油和焦炭的品質下降,影響下游利用[12-13]。含硫氣體的脫硫工藝較為成熟,可通過催化水解[14-15]、加氫水解[16-18]、濕法脫硫[19]或干法脫硫等方式得到處理[20-21],由此可知,調控煤中硫元素向氣態中遷移能夠提高煤熱解產物品質,通過成熟的含硫氣體脫硫技術,也能實現硫元素的妥善處置。因此明晰硫元素在煤熱解過程中的遷移轉化機理,進而指導煤熱解過程中硫遷移行為的調控,提高煤熱解產品的品質[22],對煤炭的高效清潔利用至關重要。
噻吩類硫化物是煤炭中重要的有機硫化合物,筆者將梳理噻吩類硫化物在煤熱解過程中的遷移轉化機理研究進展,綜述熱解溫度、升溫速率、反應氣氛、煤中雜質組分和添加劑對噻吩類硫化物遷移轉化的影響機制,以期為煤炭高效清潔熱解技術的革新與開發提供參考。
1 噻吩類硫化物的賦存與析出
1.1 噻吩類硫化物在煤炭中的賦存
硫元素在煤中的賦存形態分為無機硫和有機硫,無機硫主要包括硫化物和硫酸鹽,以及微量的單質硫[23];有機硫主要包括噻吩、硫醇、硫醚、砜和二硫化物等[24-26]。噻吩類硫化物在煤中的賦存形式主要包括噻吩、苯并噻吩、二苯并噻吩、苯并萘并噻吩以及帶有甲基等支鏈的取代物[27],這些噻吩類結構通常以橋鍵鍵合或通過弱相互作用與有機基質結構結合賦存于煤中,此外還有極少量的噻吩類硫化物以游離態存在[28-29]。由表1可以看出,噻吩類硫化物是煤中主要的含硫有機物,煤化程度越高,噻吩類硫化物含量越高[30-32]。高連芬等[33]研究表明,煤化程度低的褐煤中硫形態結構以脂肪族、芳香族硫化物為主。ZHAO等[34]發現在煤化程度較高的煙煤中,含硫多環芳烴在多環芳香族化合物中的占比為71.79%~89.22%。煤化程度較高的煤炭中噻吩硫結構通常與芳香結構相結合,以多環含硫芳烴的形式賦存[35-36]。

表1 噻吩類硫化物含量的研究
1.2 噻吩類硫化物的析出
煤熱解過程中,噻吩類硫化物的析出主要有2種方式:一是熱解過程中大分子裂解析出的噻吩類硫化物;二是煤中有機硫和無機硫轉化形成的噻吩類硫化物。
煤大分子結構在熱解過程中發生裂解,從而暴露出原本賦存其中的含硫有機結構,噻吩類硫化物隨即析出;同時極少量的游離態噻吩類硫化物也隨著大分子結構的破壞揮發析出[29]。
此外,在煤熱解過程中,無機硫會向有機硫轉化,而部分有機硫則會轉化為噻吩類硫化物。ZHANG等[40]發現褐煤熱解過程中,以黃鐵礦(FeS2)和硫酸鹽所代表的無機硫會轉化成有機硫,以更加穩定的形式固定在半焦中。李斌等[41]指出熱解環境中缺少H原子時,單質硫和FeS2會轉化成穩定的有機硫化物。能夠轉化為噻吩類硫化物的有機硫物質包括硫醚和硫醇[42],其首先轉化為含硫自由基,再與碳氫自由基聚合形成噻吩類硫化物[43-45]。前人在苯硫醇和苯硫醚的熱解產物中檢測到了苯并噻吩、二苯并噻吩等噻吩類硫化物[46]。噻吩硫析出量和有機硫含量呈正相關也間接證明了硫醇和硫醚向噻吩硫的轉化[47]。LING等[48]基于密度泛函理論(DFT)計算研究了芳香族硫醇化合物向噻吩類硫化物轉化的反應路徑,苯硫醇首先通過氫遷移和成橋環反應形成1,4-硫橋化合物,之后發生C—C鍵的斷裂形成噻吩。
綜上,噻吩類結構通常以橋鍵鍵合或通過弱相互作用與有機基質結構結合賦存于煤中,噻吩類硫化物含量會隨著煤階升高而增多;熱解過程中的噻吩類硫化物主要來源于煤中大分子結構熱裂解釋放,部分來源于無機硫和硫醚、硫醇等有機硫的轉化。
2 噻吩類硫化物熱解機理
2.1 噻吩類硫化物熱穩定性
噻吩類硫化物是一種極為穩定的有機硫化物,通常需800 ℃甚至更高才會發生分解,相關研究見表2。

表2 噻吩類硫化物熱解穩定性研究
GUO等[49]對6種含硫化合物進行熱解實驗發現,噻吩類硫化物通常比硫醇、硫醚等有機硫化物的熱解溫度更高。LIU等[50]對2-甲基噻吩、苯并噻吩和二苯并噻吩進行熱解,發現噻吩類硫化物的結構越復雜,分解所需的溫度越高。DFT計算也證實了噻吩類硫化物的高熱穩定性,表3列舉出噻吩類硫化物的起始熱解反應,這些反應的發生均需克服較高的能壘(約300 kJ/mol),且結構越復雜(如苯并噻吩、二苯并噻吩)需要克服的能壘越高。由此可知,噻吩類硫化物具有較好的熱穩定性,且結構越復雜,穩定性越好,其中帶有芳香族結構的噻吩類硫化物最難分解。
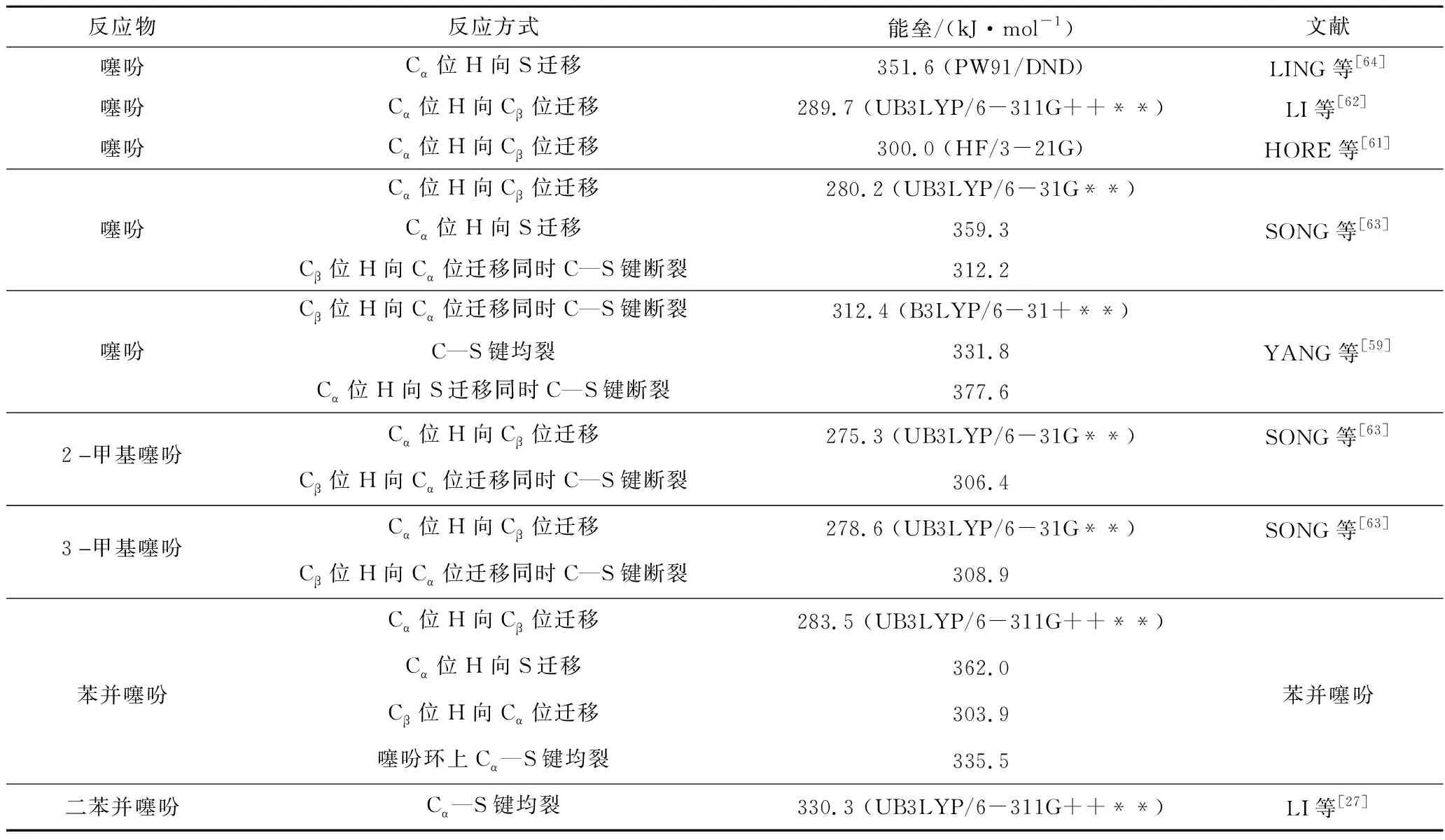
表3 基于DFT計算的噻吩類硫化物起始分解反應及能壘分析
噻吩類硫化物中的硫原子和鄰位碳原子的π電子結合會產生共軛效應,導致噻吩具有穩定的五元環結構。張福荔[51]對2-甲基噻吩感應電流密度研究表明,噻吩環外廓具有連續的感應閉合環電流,證明2-甲基噻吩的整體芳香性。多環噻吩類硫化物的硫原子也會與相鄰的苯環結構產生共軛效應形成大π鍵,因此噻吩類硫化物具有較高的熱穩定性[52-53]。
2.2 噻吩類硫化物熱解機理
噻吩類硫化物熱解過程十分復雜,通過多條反應路徑形成各種產物。
引發噻吩類硫化物分解的初始反應主要有C—S鍵均裂和氫遷移2種模式。C—S鍵均裂會直接導致噻吩環開裂形成自由基片段[58-59],反應過程沒有過渡態;而氫遷移過程涉及反應過渡態,且通常不會直接形成自由基結構。噻吩在惰性氛圍下熱解實驗結果表明C4H3和C4H4是主要的烴類產物,基于此,MEMON等[60]指出噻吩熱解由C—S鍵的均裂引發。HORE等[61]檢測到CO和丙二烯、烯酮和乙炔的存在,并提出氫遷移反應引發了呋喃的分解反應路徑;由于噻吩和呋喃具有相似的環狀結構,推測噻吩的初始分解由氫遷移反應引發。DFT計算結果表明,相比于Cβ位H原子,Cα位H原子通常更易作為初始氫遷移反應的氫供體,遷移至Cβ位[27,62-63]或S原子[64],形成Cα位具有一對孤對電子的噻吩環結構。由表3可知,C—S鍵均裂所需克服的能壘要高于Cα位H原子遷移至Cβ位的氫遷移反應。值得注意的是,初始氫遷移反應也可能導致C—S鍵斷裂。YANG等[59]基于DFT計算發現在Cα位H遷移至S原子和Cβ位H遷移至Cα位的過程中,C—S鍵的Mayer鍵級強度降低,認為在氫遷移過程中發生了C—S鍵斷裂。不同的是,SONG等[63]的DFT計算結果表明,噻吩、2-甲基噻吩和3-甲基噻吩熱解過程中只有Cβ位H遷移至Cα位時才會發生C—S鍵的斷裂。
在噻吩類硫化物熱解過程中會形成多種含硫自由基碎片,包括S自由基、SH自由基和CS自由基等[65-67]。研究認為這些含硫自由基碎片對整個熱解過程至關重要[68],SH自由基和S自由基能夠與H自由基結合形成H2S,其中H自由基可以來源于煤炭熱解,也可以來源于外部氫源(H2或水蒸氣)[69];CS自由基能夠和S自由基結合形成CS2;這些含硫自由基也會與熱解環境中的含氧基團相互結合生成硫氧化物,如SO2和COS[59,62,67]。噻吩類硫化物的熱解實驗表明H2S,COS,SO2是主要的含硫氣體產物,且通常H2S含量最高[70-71]。YAN等[66]對2-甲硫基噻吩進行了TPD-MS熱解實驗,結果顯示H2S的釋放強度最高,分別為COS和SO2釋放強度的14倍和8倍。GUO等[49]對3種噻吩類硫化物在Ar氣氛下進行了Py-GC熱解實驗,結果表明,2-甲基噻吩熱解釋放的含硫氣體中H2S的釋放強度分別是COS和SO2釋放強度的8倍和3倍;同樣地,苯并噻吩熱解釋放強度最高的含硫氣體也為H2S,是COS和SO2的10倍和5倍;而二苯并噻吩熱解釋放的H2S強度是SO2釋放強度的9倍,是COS釋放強度的近百倍。上述結果表明噻吩類硫化物結構越穩定越容易在熱解過程中生成更多的H2S。此外,由于SO2,COS的生成與含氧基團有密切關系,所以含氧量較高的環境會極大促進這2種氣體的生成[49,71]。研究表明只有在絕氧熱解環境中才能檢測到噻吩分解形成CS2產物[60],當熱解環境中存在氧元素的時候,CS2極易被含氧基團氧化為COS[72]。
為解析噻吩類硫化物熱解的氣相遷移路徑,學者們針對多種噻吩類硫化物模型開展了DFT計算,研究主要含硫基團的演化路徑[59,66,69]。圖1總結了不同DFT計算中噻吩類硫化物熱解的優勢路徑,其中,S自由基和H2S的形成通常需要經過具有巰基官能團的硫醇中間體。值得注意的是,多數學者的計算結果中,乙烯硫酮(C2H2S)是一種重要的含硫產物,但由于其化學性質活潑并未在實驗中檢測到。
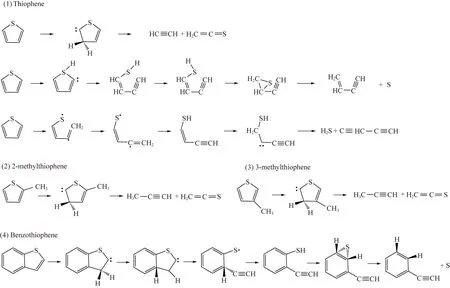
圖1 DFT計算噻吩類硫化物熱解優勢路徑[58-59,62-63]Fig.1 Dominant pyrolysis pathways of thiophene compounds based on DFT calculations[58-59,62-63]
在熱解過程中,噻吩類硫化物會分解形成含硫自由基、烷烴片段等,這些中間體能夠互相結合發生縮合反應,或與芳香結構結合最終以多環含硫芳烴的形式保留在焦油和焦炭中[43]。YAO等[73]發現噻吩類硫化物熱解生成的焦油中,二苯并噻吩和苯并萘并噻吩等分子量相對較小的噻吩類硫化物是主要的含硫產物;而分子量更大的噻吩類化合物則通常被固定于焦炭中[73-74]。基于噻吩熱解產物中含有苯并噻吩、萘、苯基噻吩的實驗結果[75],ZHANG等[76]提出噻吩極有可能通過Diels-Alder反應發生聚合,該反應過程中兩分子的噻吩聚合生成苯并噻吩,兩分子的苯并噻吩反應生成苯并萘并噻吩,由原來的單環或雙環結構變為多環含硫芳烴。此外,雙噻吩砜的熱解計算結果表明這種反應方式在動力學和熱力學上都占據優勢[51],說明Diels-Alder反應在噻吩類硫化物熱解向液相或固相遷移轉化過程中具有重要作用[43]。
基于上述研究,煤熱解中噻吩類硫化物的遷移轉化路徑如圖2所示。噻吩類硫化物主要來源于煤中大分子的分解析出以及其他有機硫和無機硫的轉化。噻吩類硫化物首先經氫遷移和C—S鍵均裂反應分解產生S自由基、SH自由基、CS自由基等自由基和含硫基團、烷烴基團,其一部分相互結合,一部分與熱解環境中的H自由基及含氧基團結合,最終形成H2S,COS,SO2,CS2等氣體逸出,其他基團會相互結合或與芳香結構發生聚合反應形成分子量更大的多環含硫芳烴化合物,最終保留在焦油或焦炭中。
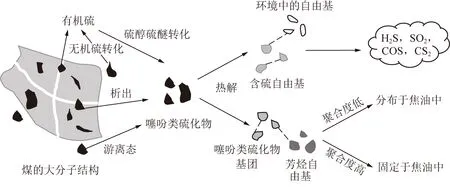
圖2 煤熱解過程中噻吩類硫化物的遷移轉化路徑Fig.2 Migration and transformation pathways of thiophene compounds during coal pyrolysis
綜上,噻吩類硫化物的共軛結構使其具有良好的熱穩定性,因此其分解需要較高的反應溫度;實驗和計算結果表明,噻吩類硫化物的分解是由氫遷移和C—S鍵斷裂所引起的,分解形成的含硫自由基與熱解環境中的氫自由基或含氧基團結合形成多種含硫氣體,另一部分含硫基團會互相結合或與烷烴基團結合形成多環芳烴結構保留在焦油和焦炭中。
3 熱解條件對噻吩類硫化物遷移轉化的影響
噻吩類硫化物熱解過程中,熱解溫度、升溫速率、反應氣氛、煤中雜質組分和添加劑等都會影響噻吩類硫化物熱解反應路徑的競爭性,進而影響產物分布。
3.1 熱解溫度和升溫速率的影響
由于噻吩類硫化物的熱穩定性較高,熱解溫度和升溫速率對其分解影響顯著。在煤熱解過程中,隨著熱解溫度的升高,脫硫率逐漸升高,脫硫效果更好[11,54]。LIU等[77]在500 ℃和700 ℃對遵義煤進行熱解,發現700 ℃下能夠使噻吩類硫化物及其他穩定的有機硫化物發生分解。馬玉川等[78]發現褐煤在500~700 ℃內熱解時,焦炭中噻吩類硫化物等有機硫含量較低,有機硫占焦炭的0.65%(質量分數),然而當熱解溫度從700 ℃升至800 ℃,焦炭中噻吩類硫化物等有機硫含量增加,達0.89%,筆者推測這可能是由于活性氫自由基的缺乏導致含硫自由基與堿性礦物質或有機質結合最終滯留在半焦中。此外在700 ℃下考察了10,20,30,40 ℃/min升溫速率對硫遷移的影響,結果表明升溫速率越慢越有利于煤中噻吩類硫化物等含硫物質的釋放,在升溫速率為10 ℃/min條件下有機硫的脫除率最高,為63.7%[78]。YU等[79]對含有大量噻吩類硫化物的焦炭在10,30,50 ℃/min升溫速率下進行熱解,發現在10 ℃/min升溫速率下脫硫率最高,達42%。但何玉遠[80]對生物質與煤的混合物(質量比3∶2)進行慢速升溫熱解,考察了升溫速率分別為5,10,15 ℃/min條件下的脫硫效果,發現升溫速率為15 ℃/min時總脫硫率最高,為87.21%,焦炭中含有的噻吩類硫化物最少。此外,升溫速率對含硫氣體析出速率有影響,較低的升溫速率(10 ℃/min)會導致含硫氣體析出速率大幅降低,因此,較長的停留時間是較低升溫速率下脫硫率更高的重要原因。
3.2 反應氣氛的影響
3.2.1 H2氣氛
H2能夠提供還原性氣氛,促進噻吩類硫化物分解向氣態遷移,主要以H2S形式釋放[81-82]。低階煤和中階煙煤在惰性和純H2氣氛下的熱解實驗表明,相比于惰性氣氛,在純H2氣氛下熱解得到的焦炭中噻吩類硫化物含量大幅降低,證明H2能促進噻吩類硫化物分解[30,83]。XING等[55]提出H自由基對噻吩類硫化物熱解產物的生成具有重要作用,在煤熱解過程中,H自由基不足,使噻吩結構之間發生縮合反應,最終導致噻吩類硫化物保留在焦油焦炭中;由H2解離生成的H自由基能和這部分噻吩結構發生加氫飽和反應,減少穩定噻吩類硫化物的生成量。YANG等[59]研究表明,由H2產生的大量H自由基更易和S,SH自由基結合生成H2S氣體逸出,從而促進噻吩類硫化物向氣態遷移轉化。MORALES等[84]發現熱解過程中產生的H自由基會直接攻擊S原子,降低噻吩分解的反應能壘,同時提出H2分子可通過加成反應形成飽和鍵來促進噻吩類硫化物分解。
3.2.2 水蒸氣氣氛

3.2.3 CO2氣氛
作為一種氧化性氣氛,CO2會對煤熱解過程中硫遷移產生影響[89]。在含有10%,20%,40%,80%(體積分數) CO2的N2氣氛下,原本向焦炭中轉移的硫元素能夠以含硫氣體形式釋放,從而提高脫硫率[90-91]。對比噻吩類硫化物在惰性氣氛和純CO2氣氛下的熱解實驗發現,CO2氣氛能夠促進噻吩類硫化物向氣態遷移,其中,促進了COS的生成,H2S和SO2的生成量被小幅抑制[50,71,92]。GUO等[71]對比了6種含硫有機化合物在Ar和純CO2氣氛下的熱解反應,發現2-甲基噻吩、苯并噻吩和二苯并噻吩這3種噻吩類硫化物都在CO2氣氛下表現出更低的分解溫度,釋放出更多含硫氣體。YANG等[93]得到了類似的實驗結果,相比于Ar氣氛,在純CO2、含有85% CO2的Ar和含有75% CO2的Ar氣氛下,產物中噻吩類硫化物明顯減少,證實CO2對噻吩類硫化物分解的促進作用。有關CO2促進噻吩類硫化物熱解的機理,前人研究[71,92,94]認為CO2的氧化性能夠降低C—S鍵的斷裂難度,從而促進噻吩類硫化物分解,在高溫下這些噻吩類硫化物會釋放出更多的CO,由此形成更多COS氣體。CHEN等[95]基于反應力場分子動力學,對CO2促進噻吩類硫化物分解的機理進行探究,表明CO2作為反應物能與噻吩類硫化物反應生成SCO2自由基,從而促進COS氣體形成,還會促進CHO和R—O自由基的形成,從而促進H2S和SO2的生成。
3.3 煤中雜質組分和添加劑的影響
煤中含有多種礦物雜質,煤種不同,礦物雜質種類和含量不同,對硫元素的遷移有重要影響。因此在熱解過程中加入不同性質的添加劑,作為反應物或催化劑來影響噻吩類硫化物的熱解[96-97],改善焦油焦炭的品質。
GU等[94]發現煤中的高嶺土雜質對熱解過程中硫的遷移具有催化作用,使得煤在惰性氛圍下熱解釋放出更多的揮發性噻吩類硫化物,減少了噻吩類硫化物向焦油和焦炭中的遷移。LIU等[98]對高鈣褐煤進行了熱解實驗,發現煤中含有的鈣幾乎完全消除了焦炭中的噻吩類硫化物,并將這些有機硫轉化為CaS。
在煤熱解過程中,多種因素都會影響噻吩類硫化物的遷移轉化。熱解溫度的提升通常能促進噻吩類硫化物的熱解,升溫速率越慢噻吩類硫化物的脫除越徹底;H2、水蒸氣和CO2氣氛能通過提供自由基和參與加成反應的方式促進噻吩類硫化物分解;煤中高嶺土能促進噻吩類硫化物的釋放,鈣能促進噻吩類硫化物轉化為CaS;添加劑會充當反應物或催化劑促進噻吩類硫化物的熱解。
4 結語和展望
筆者從噻吩類硫化物的賦存、析出和熱解3方面對噻吩類硫化物的遷移轉化機理進行綜述。系統總結了噻吩類硫化物的遷移轉化路徑,煤熱解過程中噻吩類硫化物來源于煤中大分子的分解析出以及其他硫化物的轉化,其在熱解過程中能產生含硫自由基,與熱解環境中的H自由基或含氧基團結合形成含硫氣體逸出,其他噻吩硫結構相互聚合或與芳香結構結合形成分子量更大的多環含硫芳烴,最終聚集在焦油和焦炭中。概述了熱解溫度、升溫速率、反應氣氛、煤中雜質組分和添加劑對噻吩類硫化物熱解的影響特性和機理,溫度升高能夠促進煤中噻吩類硫化物的脫除;升溫速率越慢,噻吩類硫化物脫除率越高;H2、水蒸氣和CO2氣氛通過提供自由基、參與加成反應等方式促進噻吩類硫化物分解;煤中的高嶺土能促進噻吩類硫化物釋放,鈣質礦物能與噻吩類硫化物反應向CaS轉化;不同性質的添加劑會充當反應物或催化劑對噻吩類化合物的熱解反應產生不同影響。
為提高脫硫效果和熱解產物品質,需對煤熱解過程中噻吩類硫化物遷移進行干預調控。由噻吩硫化物的遷移轉化機理可知,適當提高熱解溫度、降低升溫速率、改變熱解氣氛可定向促進噻吩類硫化物的氫遷移反應和C—S鍵斷裂,促進噻吩類硫化物分解向氣相遷移;改變熱解氣氛或加入添加劑增加熱解環境中的氫自由基和含氧自由基團,能促進噻吩類硫化物基團與其結合,減少多環含硫芳烴化合物的生成。
在今后的研究中,仍需進一步明晰噻吩類硫化物遷移至液相和固相中的路徑,探究反應器型等多種熱解條件耦合作用下噻吩類硫化物的熱解特性,為煤炭熱解清潔利用技術開發提供重要的理論基礎。
