奶牛乳脂代謝關鍵miRNAs的篩選及鑒定
2023-01-03 02:28:30劉佳敏禹保軍馮小芳顧亞玲
畜牧獸醫學報 2022年12期
劉佳敏,禹保軍,母 童,張 迪,馮小芳,張 娟,王 影,溫 萬,顧亞玲*
(1.寧夏大學農學院,銀川 750021;2. 寧夏回族自治區反芻動物分子細胞育種重點實驗室,銀川 750021;3.寧夏畜牧工作站,銀川 750002)
牛乳中富含多種營養物質,蛋白質、脂肪、微量元素以及多種維生素是人體必不可少的營養來源,其中脂肪和蛋白質含量是評價乳品質的重要參考因素,產奶性狀亦是奶牛育種的主要目標[1]。近年來在科學育種及飼喂下,奶牛產奶量已大幅提高,但乳脂率增長甚微。甘油三酯、膽固醇和游離脂肪酸是構成乳脂的主要成分,乳脂是乳的重要組成部分,在乳品質評價中起到決定性作用。奶牛生長發育過程中乳脂合成代謝受多因素的影響,其性狀表現與遺傳因素密切相關,受到多個轉錄因子的調節[2]。而目前對于奶牛乳脂代謝分子機制的相關研究還十分欠缺,尤其是對高低乳脂荷斯坦奶牛乳脂合成的轉錄后調控機制方面的研究。因此,利用分子遺傳學理論和技術方法研究影響奶牛乳脂性狀的調控因子,以期獲得調控奶牛乳脂率的關鍵mRNA和miRNA并驗證其生物學功能,通過遺傳育種手段提高奶牛的泌乳性能,對于奶牛的育種工作具有重要意義。
非編碼RNA(ncRNA)作為基因表達的一種重要調控因子,在農業生產的多個領域被廣泛研究。其中,microRNAs(miRNAs)是一類長度約22 nt左右的內源性小非編碼RNA,通過堿基互補配對方式與mRNA非編碼區結合位點結合,調控靶基因降解或抑制轉錄后翻譯過程,從而調控基因的表達[3]。已有研究闡明,乳脂合成的主要方式包括脂肪酸從頭合成和血液中長鏈脂肪酸的攝取[4]。隨著生物學技術的發展,已挖掘出一系列調控乳脂合成代謝的主效基因與轉錄因子。在調控乳脂代謝方面,催乳素作為泌乳的關鍵激素可以促進miRNA-23a、miRNA-27b、miRNA-103和miRNA-200a表達,而且miRNA-200a過表達后可以抑制脂滴形成相關基因的表達[5]。microRNA-193a-5p[6]和bta-miR-34b[7]均會降低甘油三酯和不飽和脂肪酸含量,證實在脂質代謝中的關鍵作用。在調控乳脂合成方面,過表達bta-miR-125a[8]和miR-103[9]能夠促進乳脂合成相關基因的表達,增加甘油三酯的積累、脂滴的形成和不飽和脂肪酸的比例。此外,miRNA作為生物性狀功能發揮重要的轉錄因子,可以影響乳腺上皮細胞中的脂質含量[10],也可以通過多種代謝通路的調節促進甘油三酯和膽固醇的合成,是闡明乳脂性狀表現的關鍵特征分子[8,11]。
本研究篩選乳脂率具有極端差異的荷斯坦奶牛,基于轉錄組測序鑒定高低乳脂荷斯坦奶牛乳腺上皮細胞(bovine mammary epithelial cells, BMECs)中乳脂代謝的關鍵miRNA。通過miRNA-mRNA關聯分析確定互作網絡中的核心差異表達miRNA和mRNA,并驗證其在高低乳脂荷斯坦奶牛乳腺上皮細胞中的表達量,分析其與乳脂率的相關性,為從轉錄水平探究寧夏地區荷斯坦奶牛乳脂合成代謝的調控機制提供理論依據。
1 材料與方法
1.1 試驗材料
本試驗選取寧夏農墾賀蘭山奶業茂盛牧場的健康中國荷斯坦奶牛,飼養背景一致(表1),月齡相近,處于泌乳中后期(150~220 d)的第一胎次荷斯坦牛400頭。采集每頭牛早、中、晚班次的乳樣進行DHI測定,篩選體細胞數在10萬·mL-1以內,乳脂率具有極端差異的荷斯坦奶牛(表2)[12]各4頭。采集牛奶并裝入50 mL離心管中,將離心管封口并裝入加有溫水的保溫瓶中,保溫瓶溫度始終保持在37 ℃,帶回實驗室進行乳腺上皮細胞的分離培養[12]。
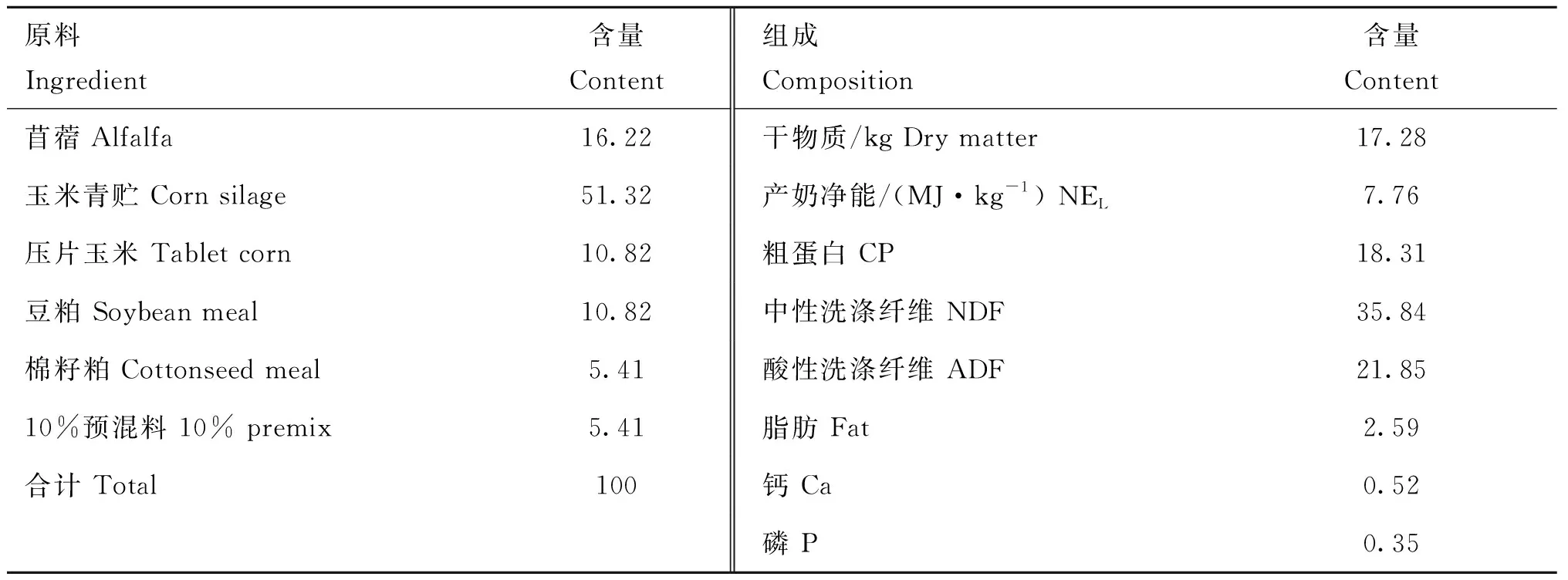
表1 日糧成分及營養成分(以干物質為基礎)百分比
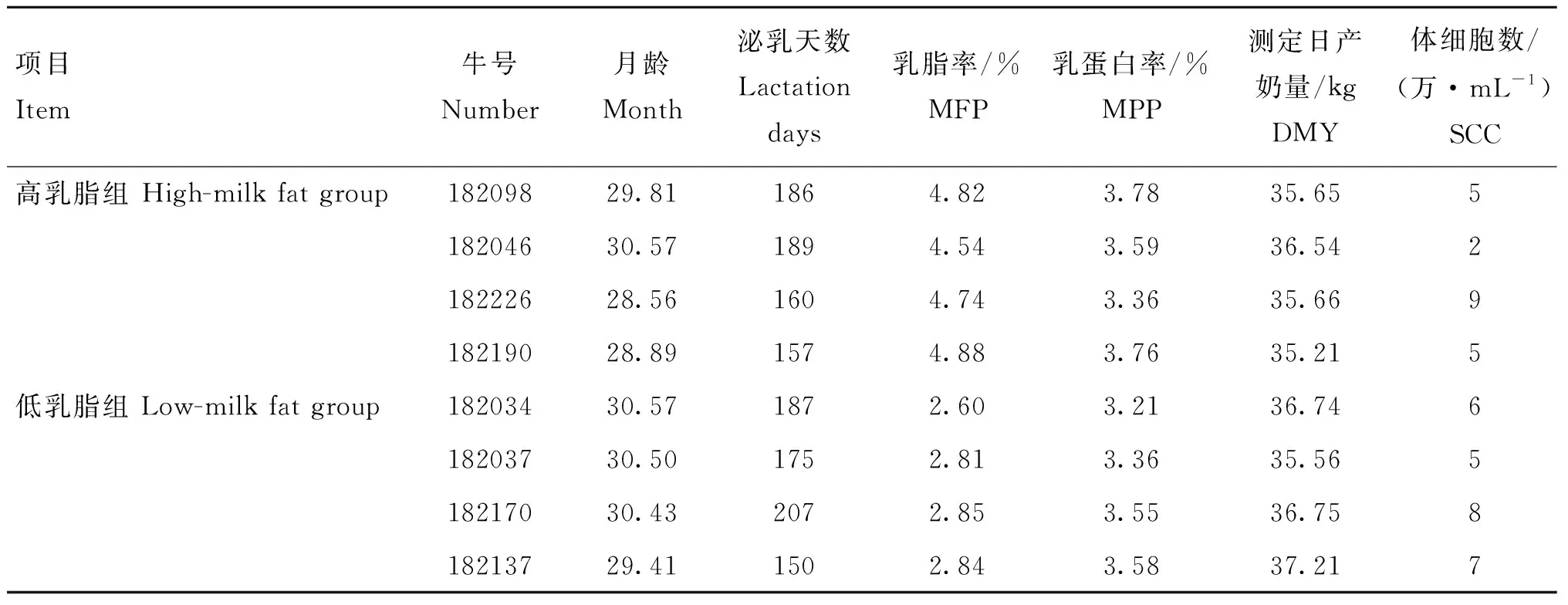
表2 荷斯坦牛高低乳脂率和SCC
1.2 方法
1.2.1 乳腺上皮細胞總RNA的提取與質量檢測 TRIzol法提取高低乳脂奶牛乳腺上皮細胞的總RNA。根據10 g·L-1瓊脂糖凝膠電泳條帶判斷RNA有無降解和污染(圖1),用分光光度計和Qubit?RNA分析試劑盒檢測RNA的濃度和純度,使用RNA Nano 6000檢測試劑盒在Bioanalyzer 2100系統上檢測RNA的完整性。乳腺上皮細胞總RNA的OD值(OD260nm/OD280nm比值)均在1.8~2.2之間,RIN值(完整指數)均大于8.0,說明RNA純度和完整性都較好。
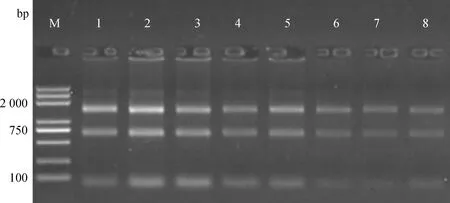
M.DNA相對分子質量標準;1~8. 依次為樣本H_2190、H_2226、H_2098、H_2137、L_2046、L_2170、L_2034和L_2037M.DL2000 DNA marker; 1-8. The samples H_2190, H_2226, H_2098, H_2137, L_2046, L_2170, L_2034 and L_2037圖1 提取的乳腺上皮細胞總RNA的瓊脂糖凝膠電泳Fig.1 Agarose gel electrophoresis of total RNA extracted from mammary epithelial cells
1.2.2 文庫構建與轉錄組測序 乳腺上皮細胞總RNA提取成功并檢測合格后,以每個樣品3 μg總RNA為起始量。由于small RNA的5′端具有完整的磷酸基團,3′端具有羥基,這個特殊結構可以直接加接頭,進行反轉錄cDNA的合成。然后對cDNA片段純化、PCR擴增、PAGE富集篩選目標片段、切膠回收獲得cDNA文庫。庫檢合格后,通過Illumina-Hiseq對不同樣品的cDNA文庫按照有效濃度及目標下機數據量的需求進行測序,由北京諾禾致源科技股份有限公司完成。
1.2.3 測序數據處理及差異miRNA篩選 測序得到的原始圖像數據文件中包含reads信息及其對應測序質量情況。為了保證數據分析的質量,需對Raw data中堿基測序錯誤率(Qphred =-10lg e)、帶接頭、低質量的reads進行處理,分析Q20,Q30和GC含量,即最后獲得的clean reads質量較高。測序結果質量控制之后,篩選8個樣品中small RNA(sRNA)的長度,用bowtie將長度在18~35 nt的sRNA與牛(Bostaurus)參考基因組進行比對。將比對上的reads進一步匹配miRBase中已有的序列,可得到已知miRNA的二級結構和長度等信息。剩下的未匹配的reads借助miRNA前體的發夾結構,使用miREvo和miRdeep2軟件預測樣品中novel miRNAs。對于已知和novel sRNA中的其他各類型RNA,應當通過技術手段注釋并去除。基于高乳脂和低乳脂比較組間有生物學重復,采用DESeq2分析各差異表達組合,得到各樣本間基因的表達水平之后,通過相關性分析檢驗試驗樣本選擇的可靠性及合理性。用TPM歸一化處理已知和新預測miRNA的表達量,以P<0.05為標準篩選比較組間差異表達miRNA。
1.2.4 差異表達miRNA靶基因預測及功能富集分析 使用miRanda和RNAhybrid預測miRNA的靶基因,取其交集即為后續分析的候選靶基因集合。采用Goseq方法對差異表達miRNA的靶基因集合進行Gene Ontology分析(http://www.geneontology.org/)。P<0.05的GO條目可認為是差異基因顯著富集。基于KEGG(Kyoto Encyclopedia of Genes and Genomes)數據庫(http://www.genome.jp/kegg/)中的Pathway確定候選靶基因參與的主要生化代謝途徑和信號轉導途徑,以P<0.05為條件確定差異基因顯著富集的KEGG通路。
1.2.5 miRNA-mRNA關聯分析 選擇高低乳脂組差異表達miRNAs靶向的差異表達mRNAs,使用Cytoscape_v3.7.2軟件構建互作網絡。然后用pearson相關檢驗計算差異表達miRNAs及靶mRNAs與乳脂率、乳蛋白率、日產奶量和體細胞數之間的相關性,進而確定調控乳脂率的關鍵miRNA-mRNA。
1.2.6 差異表達miRNA和mRNA的RT-qPCR驗證 隨機選擇差異表達的9個miRNAs和6個mRNAs對測序數據進行驗證。miRNA的引物采用莖環法設計,mRNA的引物用Primer6.0軟件設計(表3),以U6和GAPDH分別作為miRNA和mRNA的內參。使用與轉錄組測序樣本一致的RNA,按照反轉錄試劑盒PrimeScriptTMRT reagent Kit(Perfect Real Time)的說明合成cDNA,以cDNA為模板,使用QuantiNovaTMSYBR?Green試劑盒進行實時熒光定量PCR檢測。反應體系:上、下游引物各0.5 μL,cDNA 2 μL,2× QuantiNova SYBR Green PCR Master Mix 10 μL,RNAase Free ddH2O補至20 μL。反應條件:95 ℃預變性2 min;95 ℃變性5 s,退火(退火溫度見表3)30 s,40個循環,每個樣品3個重復。利用 2-ΔΔCt計算miRNA和mRNA的相對表達量,結果表示為“平均值±標準誤”。

表3 引物信息
2 結 果
2.1 測序數據評估及樣品間相關性分析
荷斯坦奶牛乳腺上皮細胞樣本8個small RNA文庫測序獲得11 976 914~16 235 680的clean reads,占總raw reads的比例均在97%以上。各個樣品的測序錯誤率(error rate)均為0.01%,Q20和Q30的比例均在97%和91%以上,堿基GC含量基本相等,堿基組成穩定均衡(表4)。高乳脂組和低乳脂組sRNA序列與參考基因組的匹配率均在93%以上。將匹配上的reads與miBase中特定范圍序列進行比對分析,可得到8個樣本known miRNA共有21 346種、85 152 090個,novel miRNA分別為744、192、316、364、364、245、366和366(表5)。sRNA的序列長度主要集中在21~24 nt,其中23 nt的sRNA序列比率最高(圖2)。通過定量分析并用TPM歸一化處理,發現8個樣本中只有個別miRNA表達量極高,多數表達較為集中(圖3A)。為了排除異常樣本帶來的誤差,計算評估樣本間pearson相關性:R2>0.9(圖3B)。以上結果表明測序數據質量較高,可以用于后續的差異miRNA分析。
2.2 高低乳脂組奶牛乳腺上皮細胞差異表達miRNA篩選
為了探究調節奶牛乳脂率的關鍵因子,在高乳脂組和低乳脂組之間共鑒定了34個差異表達miRNAs(上調16個,下調18個)(表6,圖4A),其中33個是已知 miRNAs,1個新 miRNA。通過層次聚類分析,進一步確定高乳脂組和低乳脂組間差異表達miRNAs的表達水平有顯著差異(圖4B)。
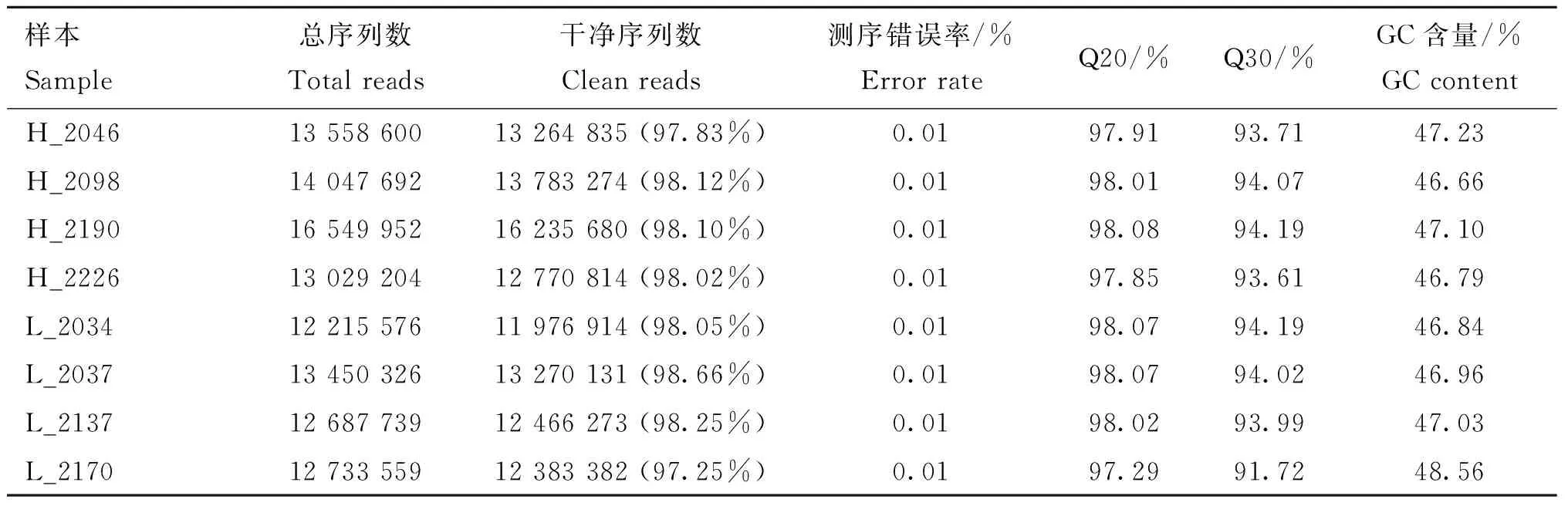
表4 原始數據質量評估
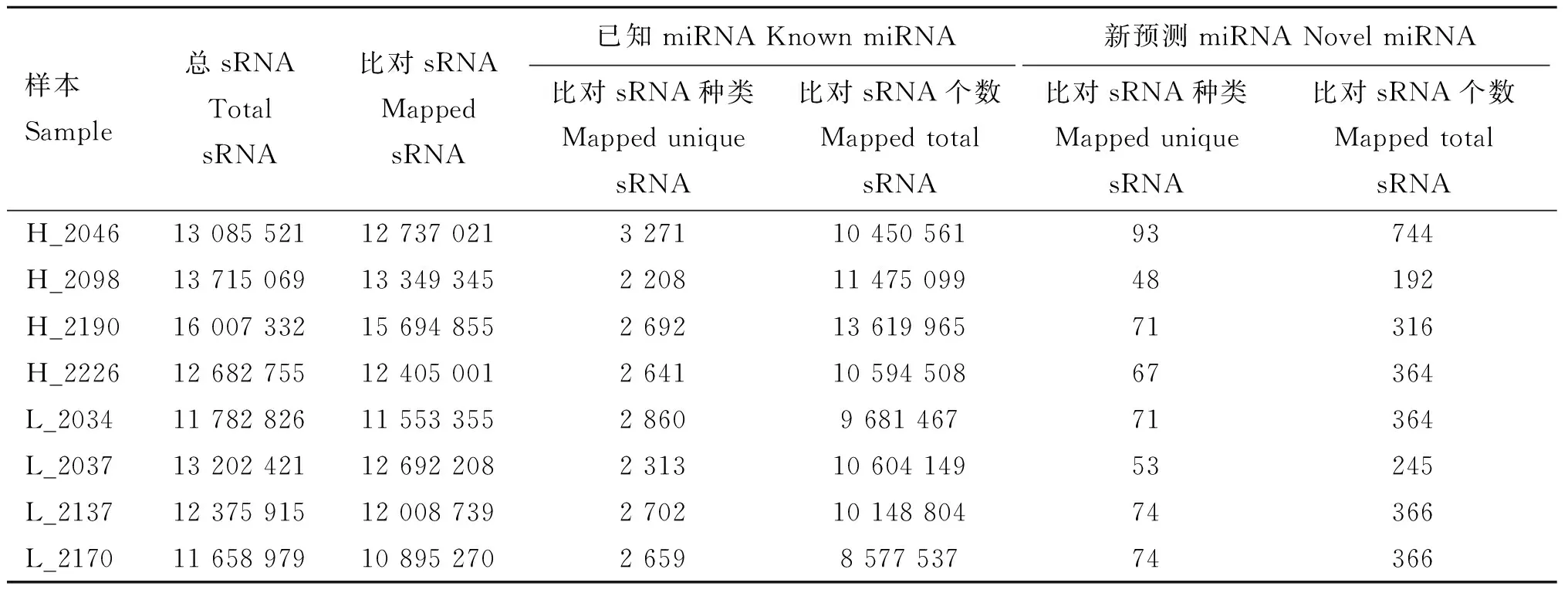
表5 sRNA與參考基因組比對情況
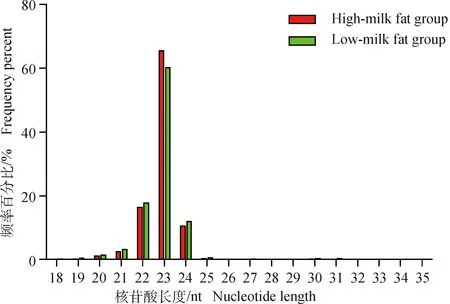
圖2 sRNA片段的長度分布Fig.2 Length distribution of sRNA fragments
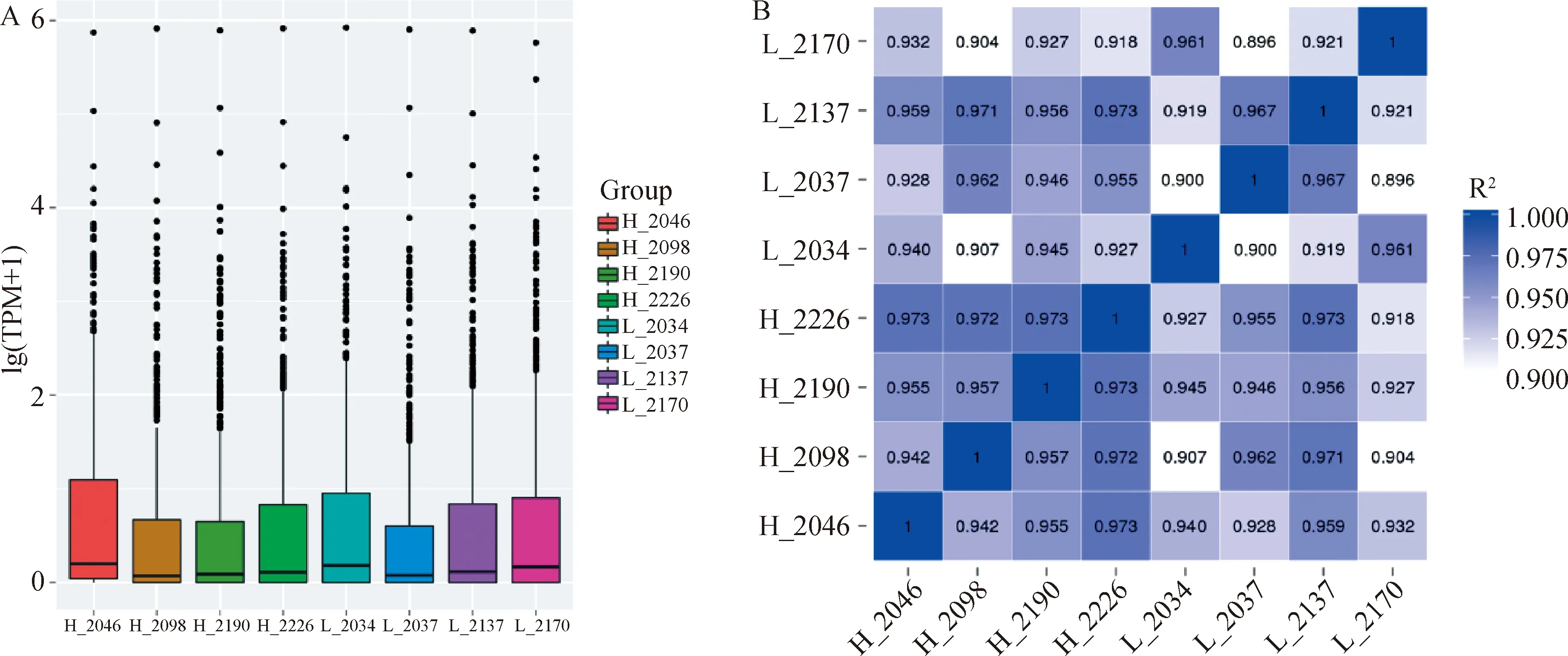
A. miRNAs的TPM分布;B. 樣品間miRNA表達量相關性A. TPM distribution of miRNAs; B. Correlation of miRNA expression between samples圖3 miRNA測序數據評估Fig.3 miRNA sequencing data evaluation
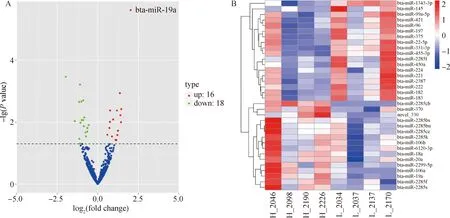
A.差異表達miRNAs火山圖; B.差異miRNAs表達譜聚類熱圖A. Volcano map of differentially expressed miRNAs; B. Heat map of clustering of differential miRNAs expression profiles圖4 差異表達miRNAs鑒定及聚類分析Fig.4 Identification and clustering analysis of differentially expressed miRNAs
2.3 差異表達miRNAs的 GO和KEGG富集分析
采用miRanda和RNAhybrid兩個軟件預測得到已知miRNA和新miRNA的靶基因,取其交集進行GO功能和KEGG通路注釋分析。結果顯示,靶基因集合顯著富集的GO條目有241個。在生物學過程(biological process,BP)中富集的203個條目包括細胞內信號轉導、單一生物體過程和細胞死亡等,分子功能(molecular function,MF)中富集的17個條目包括激酶結合、磷酸轉移酶活性和腎上腺素受體結合等,細胞組分(cellular component,CC)中富集的21個條目包括細胞質和細胞內等(圖5A)。KEGG通路富集結果顯示,差異表達miRNAs的靶基因共富集到272條通路,在15條通路顯著富集。其中TNF信號傳導途徑、MAPK信號傳導途徑、Ras信號傳導途徑、Rap1信號通路、催產素信號傳導途徑和GnRH信號傳導途徑等通路(圖5B)與乳脂代謝密切相關。結合GO和KEGG分析可發現,奶牛乳脂代謝可能受到多種機制的調節,包括信號分子傳導、能量代謝和激酶活性調節。
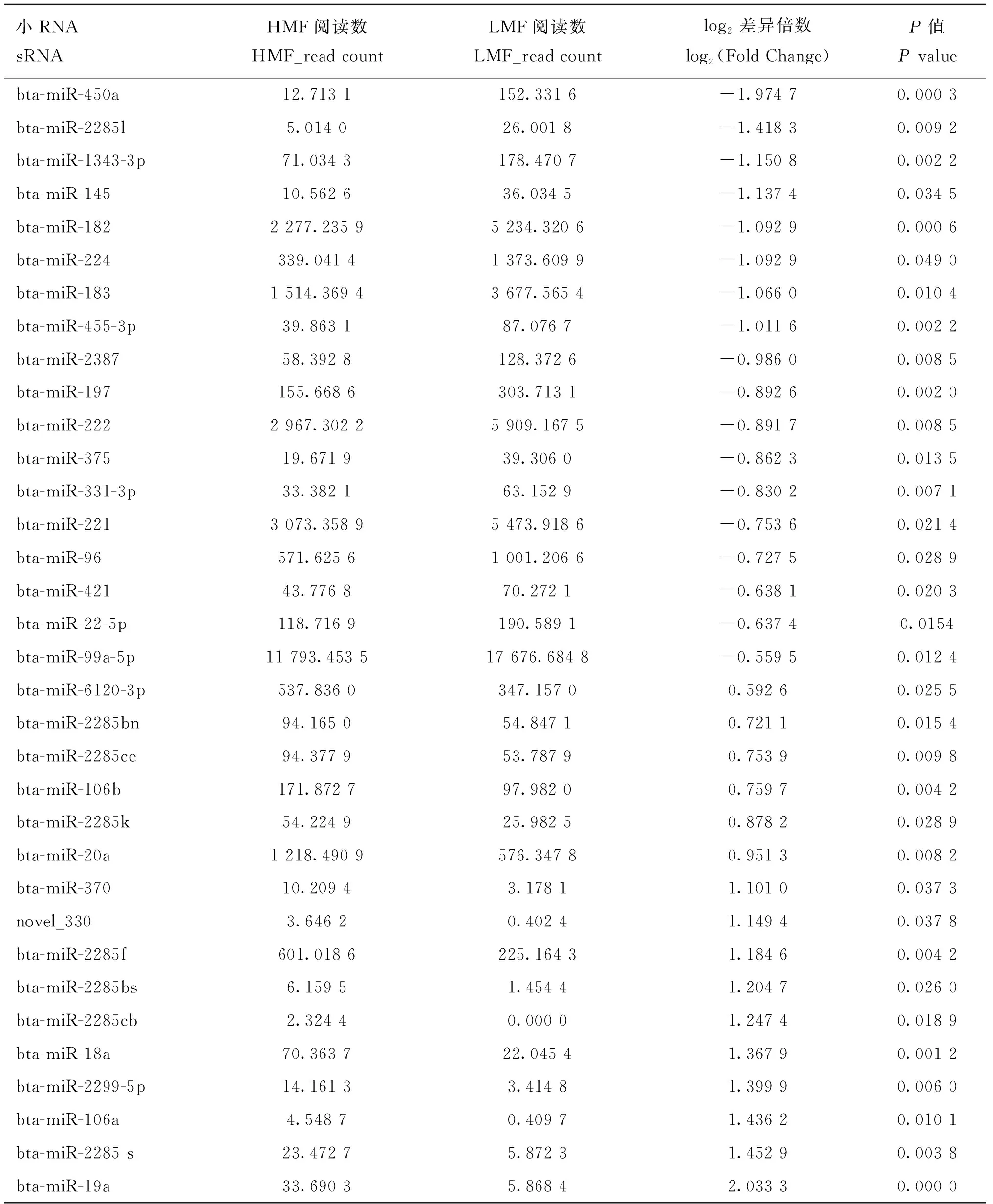
表6 差異表達miRNAs
2.4 miRNA-mRNA關聯分析
為了確定與乳脂代謝相關的關鍵miRNA-mRNA,構建差異表達miRNA與差異表達靶mRNA的互作網絡。結果共鑒定出16個miRNA-mRNA互作對(9個miRNAs和9個mRNAs),其中有8個互作對具有負調控關系(圖6A)。對9個差異靶標基因進行功能分析發現,主要富集于蛋白激酶A結合、磷脂酰肌醇磷酸酯酶活性、mRNA分解代謝和胰島素分泌的正向調節等多個生物學功能(圖6B)。只有一個基因MTM1顯著富集到磷酸肌醇的代謝和磷脂酰肌醇信號系統,與甘油三酯和乳脂合成密切相關。此外,將差異表達miRNA及靶標mRNA與乳脂率進行相關性分析,發現miR-1343-3p和MTM1與乳脂率顯著負相關,miR-370、miR-2285cb和SRRM2與乳脂率顯著正相關(圖6C)。綜上,可推測miR-370-MTM1、miR-1343-3p-DIS3L2、miR-2285cb-XLOC-122799和miR-2387-SRRM2是調控乳脂代謝的關鍵因子。
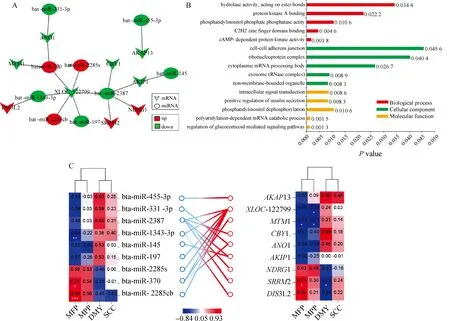
A.差異表達miRNAs與差異靶mRNAs互作網絡;B.差異靶mRNAs功能分析; C.差異miRNA-mRNA與乳成分之間的相關性A. Interaction network between differentially expressed miRNAs and differential target mRNAs; B. Functional analysis of differential target mRNAs; C. Correlation between differential miRNA-mRNA and milk components圖6 miRNA-mRNA關聯分析Fig.6 miRNA-mRNA association analysis
2.5 差異miRNAs與mRNAs的實時熒光定量PCR驗證
選擇組間差異表達的9個miRNAs和6個mRNAs驗證測序結果的可靠性。結果如圖6所示,miR-20a、miR-18a、miR-2285f、miR-106b和miR-19a在高乳脂組乳腺上皮細胞中的表達高于低乳脂組,表現為上調,miR-445-3p、miR-1343-3p、miR-224和miR-182表現為下調。DIS3L2、AKAP13、MTM1、ANO1、CBY1和AKIP1在高乳脂組的乳腺上皮細胞中的表達低于低乳脂組,均表現為下調。RT-qPCR結果與轉錄組測序結果相對表達量存在差異,但其調控方向和表達模式均一致(圖7、圖8),說明轉錄組測序結果可靠,可用于后續功能試驗研究。
3 討 論
乳腺上皮細胞是主要的產乳細胞,是研究乳脂的重要材料[13-15]。乳是由乳腺上皮細胞合成并分泌的。乳脂是乳中的重要成分之一,其合成與代謝受到嚴格的程序性調控,在生產上受到飼養管理、營養、疾病以及遺傳等多個因素的影響[16]。miRNA作為表觀遺傳調控研究的熱點,在細胞生長[17]、增殖[18]、分化[19]和凋亡[18]等多種生物學過程中廣泛研究。在產乳性狀方面,miRNA能夠調控乳腺的發育[20]、乳腺細胞的增殖和分化、乳脂代謝[21]及泌乳過程[22]。因此,本研究選擇乳脂率極端差異的荷斯坦奶牛,從牛奶中分離培養原代乳腺上皮細胞并進行轉錄組測序,以miRNA與mRNA的靶向關系為基礎進行分析,進而確定寧夏地區荷斯坦奶牛乳脂的遺傳特性及機理。
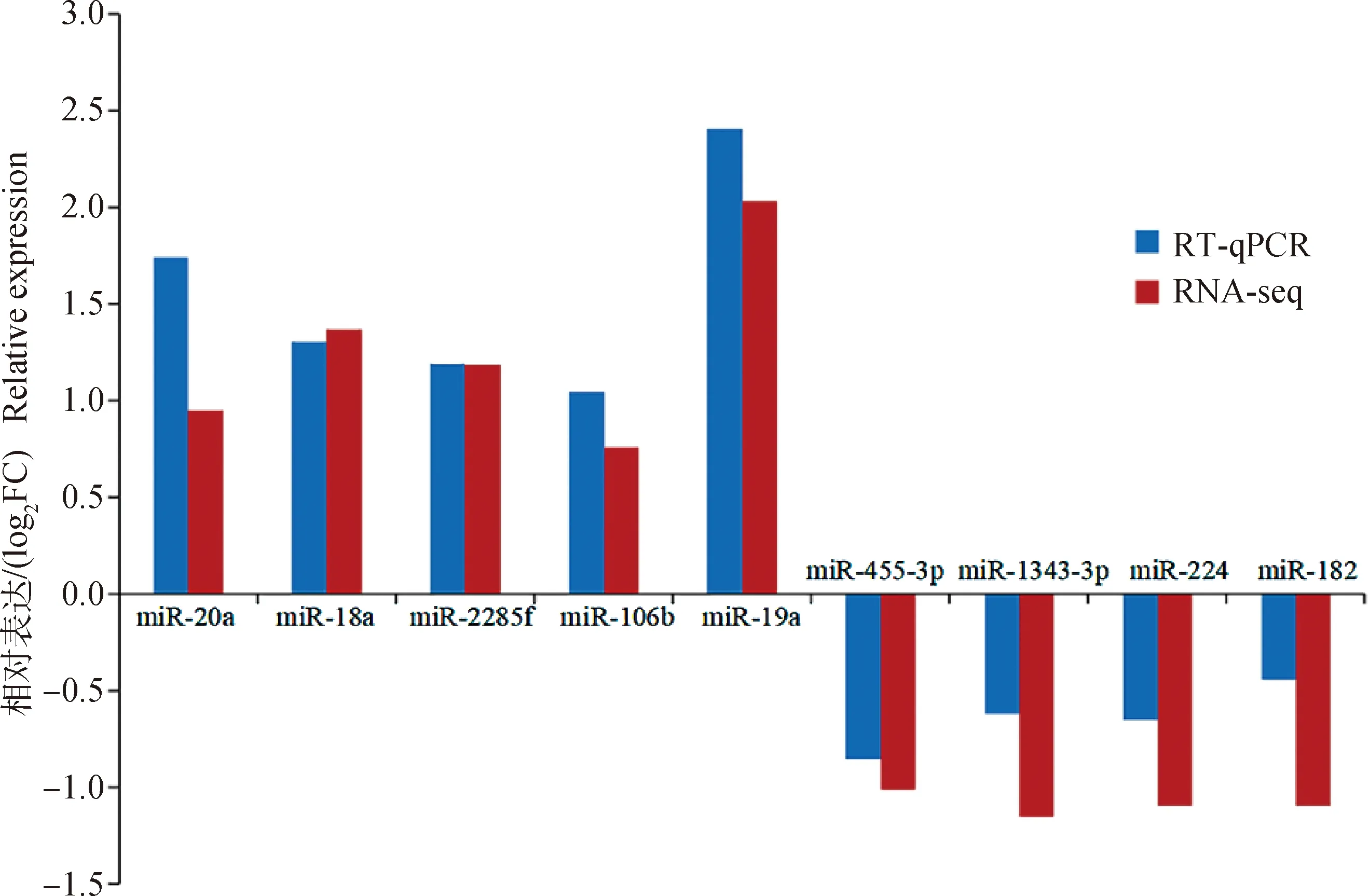
圖7 差異表達miRNAs的RT-qPCR和RNA-seq定量分析比較Fig.7 Comparison of quantitative analysis of differentially expressed miRNAs by RT-qPCR and RNA-seq
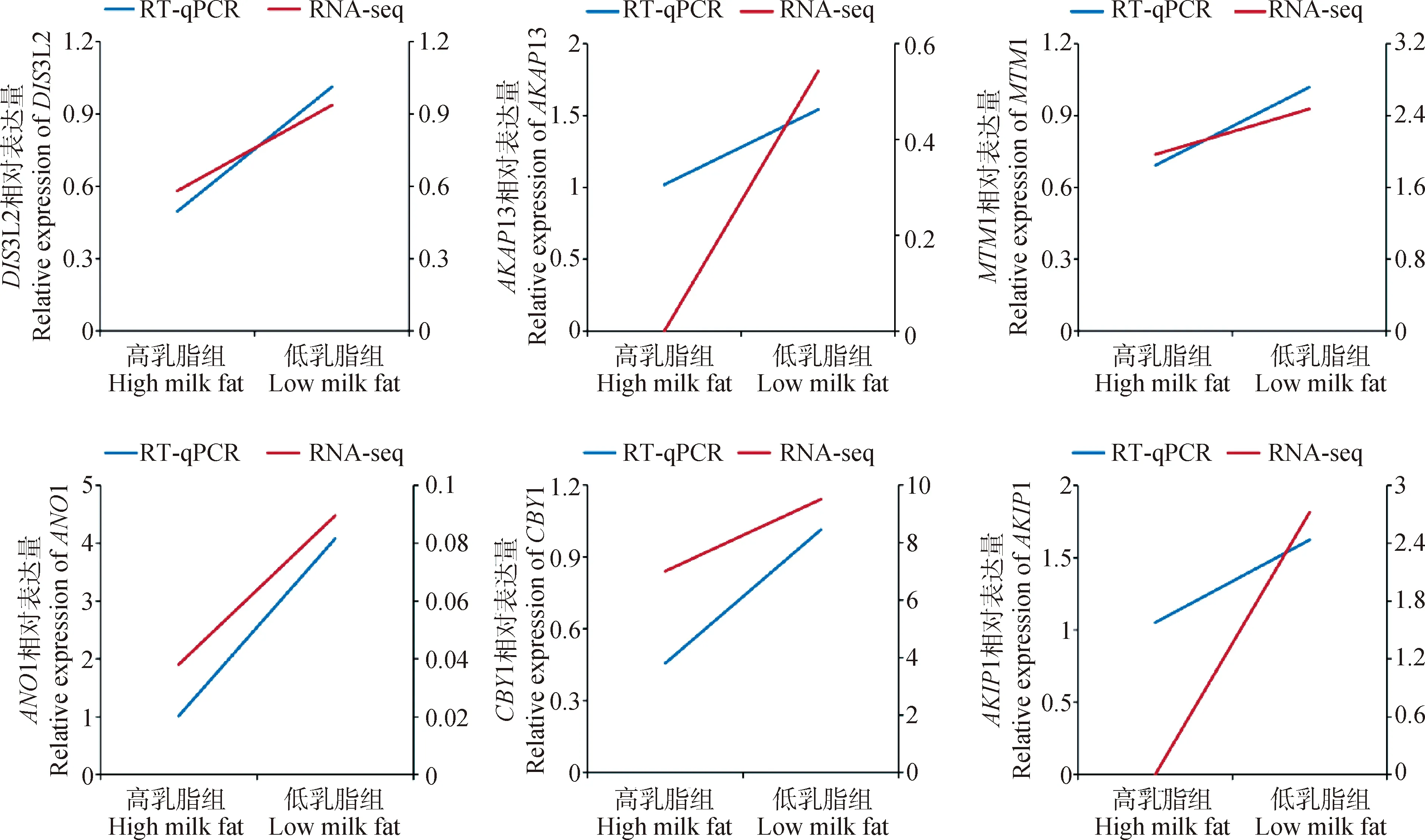
圖8 差異表達mRNAs的RT-qPCR和RNA-seq定量分析比較Fig.8 Comparison of quantitative analysis of differentially expressed mRNAs by RT-qPCR and RNA-seq
miRNA作為重要的小非編碼調控因子,可以靶向靶基因在轉錄和翻譯水平調節其表達,從而影響個體性狀表現。miR-370作為新型的脂肪生成調節劑,在脂質代謝中起重要作用。研究發現,過表達miR-370可以顯著抑制脂肪的形成和分化,導致與脂肪形成相關的基因PPARγ和aP2表達下調,進而使甘油三酯的積累量降低[23]。miR-370-3p通過靶向Mknk1對脂肪生成和脂肪酸代謝起著重要調控作用[24]。miR-197與脂肪也具有調控關系,miR-197-3p會使甘油三酯和膽固醇的水平下降,但lncRNA HCG18-miR-197-3p軸會增加脂肪積累[25]。根據miRNA-mRNA互作關系,確定可能導致相關基因表達變化的功能性miRNA[26]。本研究通過miRNA-mRNA互作分析發現,bta-miR-370、bta-miR-197與ANO1、MTM1、XLOC-122799有調控關系,可視化網絡圖中以上基因與bta-miR-331-3p、bta-miR-2387、bta-miR-1343-3p、bta-miR-2285 s和bta-miR-2285cb互作。因此,推測以上miRNAs可能通過靶向靶標基因ANO1、MTM1、XLOC-122799、DIS3L2、SRRM2、NDRG1、CBY1和AKIP1調控乳脂代謝過程。同時,結合miRNA-mRNA與乳成分相關性分析,發現bta-miR-1343-3p、bta-miR-2285cb、bta-miR-370、MTM1、SRRM2與乳脂率顯著相關。因此,可推測miR-370-MTM1、miR-1343-3p-DIS3L2、miR-2285cb- XLOC-122799和miR-2387-SRRM2是調控乳脂代謝的關鍵因子。
mRNA作為重要的調控因子直接或間接的參與乳腺的生長發育[27]、泌乳的啟動、乳脂的合成[28-29]。磷酸肌醇是脂質信號分子,其在脂質轉運中具有重要作用[30]。本研究通過靶基因富集篩選出兩條關鍵通路磷酸肌醇代謝和磷脂酰肌醇信號通路,有研究證實,磷酸肌醇代謝和磷脂酰肌醇信號通路可調節乳脂合成中的關鍵酶,參與脂類生物合成過程[31]。通過功能注釋篩選出差異表達顯著的基因共6個,A激酶錨定蛋白13(A-kinase anchor protein 13,AKAP13)、A 激酶相互作用蛋白1(A-kinase-interacting protein 1 isoform X1,AKIP1)、Anoctamin 1(ANO1)、chibby同源蛋白1(protein chibby homolog 1,CBY1)、DIS3 樣核酸外切酶 2(DIS3-like exonuclease 2,DIS3L2)和肌管蛋白(myotubularin,MTM1)在高乳脂和低乳脂荷斯坦奶牛乳腺上皮細胞中表達量較高。通過STRING數據庫分析,MTM1參與磷酸肌醇代謝和磷脂酰肌醇信號系統等多個通路,具有磷脂酰肌醇代謝過程、磷酸酶活性和脂質修飾等功能;CBY1通過與CTNNB1/β-catenin結合并通過與 TCF/LEF 轉錄因子的競爭抑制β-catenin介導的轉錄激活來抑制 Wnt/Wingless 通路,有促進脂肪細胞分化的能力。AKAP13參與MAPK、cAMP、Rap1和Wnt信號通路等多個經典通路;ANO1參與MAPK、cAMP、Rap1、PI3K-Akt 和AMPK信號通路等多個經典通路。有研究證明,PI3K-Akt、MAPK、TNF和Wnt等都是與脂代謝相關的通路[32-34],在奶牛的乳脂代謝與合成中起重要調控作用。以上結果進一步說明,MTM1、CBY1、AKAP13和ANO1可能參與調節乳脂合成代謝過程,具體調控機制有待進一步研究。
4 結 論
本研究通過RNA-seq技術描述了中國荷斯坦牛奶牛乳腺上皮細胞中的miRNA表達譜,從高乳脂和低乳脂組篩選出34個差異表達miRNAs,其中16個上調,18個下調。通過功能分析確定MAPK信號通路、Ras信號通路、Rap1信號通路、催產素信號通路和磷酸肌醇代謝等通路是乳脂代謝所必需的。通過miRNA-mRNA聯合以及乳脂率相關性分析,鑒定到miR-370-MTM1、miR-1343-3p-DIS3L2、miR-2285cb- XLOC-122799和miR-2387-SRRM2可能直接或間接參與乳脂合成代謝過程,其可作為奶牛泌乳性狀分子調控功能研究的候選基因。
猜你喜歡
音樂探索(2022年2期)2022-05-30 21:01:37
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
中國特種設備安全(2018年11期)2019-01-08 02:08:32
電子制作(2018年18期)2018-11-14 01:48:24
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
山東工業技術(2016年15期)2016-12-01 05:31:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:46
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
山東女子學院學報(2014年6期)2014-03-01 02:24:55
