去鐵胺對脂多糖誘導小鼠黑質多巴胺能神經元損傷的影響
2022-12-27 04:36:23李映慧謝俊霞王俊
青島大學學報(醫學版) 2022年3期
關鍵詞:小鼠
李映慧,謝俊霞,王俊,
(1 青島大學醫學部基礎醫學院生理學教研室,山東 青島 266071; 2 青島大學腦科學與疾病研究院)
帕金森病(PD)是一種進展緩慢的神經退行性疾病,其發病機制迄今尚不明確,許多研究表明慢性炎癥參與了PD的發病[1-2]。脂多糖(LPS)作為一種炎癥源,已被證明是實驗動物黑質紋狀體多巴胺能神經元死亡的有效引發劑[3]。C57BL/6小鼠黑質、紋狀體、側腦室內注射LPS均可以誘導黑質酪氨酸羥化酶(TH)陽性的多巴胺能神經元脫失、運動行為改變和紋狀體多巴胺減少[4]。有研究表明,黑質中鐵的異常沉積在PD的發病中起著關鍵作用[5-6]。過量的鐵可以通過Fenton反應促進羥自由基的生成,造成細胞損傷。LPS在小鼠模型中引發的神經元損傷,與腦鐵積累和一系列炎癥級聯反應有關[7]。本實驗通過黑質注射LPS及鐵螯合劑去鐵胺(DFO),采用免疫組織化學方法檢測黑質中TH陽性多巴胺能神經元的數量,探討DFO對LPS誘導的多巴胺能神經元損傷是否具有保護作用。
1 材料與方法
1.1 實驗動物及試劑
SPF級雄性C57BL/6小鼠40只,8周齡,體質量(20±2)g,購于北京維通利華實驗動物有限公司。小鼠單籠飼養于有適量食物和水的潔凈動物房內,可以自由飲食。動物房室溫(20±2)℃,濕度(50±2)%,12/12 h晝夜循環光照。小鼠適應性飼養1周進行后續實驗。動物使用和管理經青島大學動物倫理委員會批準。主要試劑:LPS、DFO購于美國Sigma公司,TH兔抗多克隆抗體購于英國Abcam公司,其余試劑均從當地試劑公司購買。
1.2 動物分組與處理
將40只實驗小鼠隨機分為對照組、LPS組、DFO組和LPS+DFO組,每組10只。小鼠用異戊烷麻醉后固定在腦立體定位儀上,調整耳桿使小鼠頭部位于同一平面。碘附消毒后剪開小鼠顱腦背側皮膚,用體積分數0.03的過氧化氫溶液擦拭顱骨表面至顱縫和前后囟清晰可見。黑質定位坐標:前囟后3.1 mm,左右旁開1.5 mm,深度4.1 mm。通過顯微注射針以0.2 μL/min的流量注射完畢后留針10 min。對照組注射生理鹽水1 μL,LPS組注射1 mg/L的LPS 1 μL,DFO組注射50 mmol/L的DFO 1 μL,DFO+LPS組注射1 mg/L LPS和50 mmol/L DFO的混合液1 μL,繼續喂養7 d進行后續檢測[8]。
1.3 TH陽性神經元檢測
采用免疫組織化學法檢測TH陽性神經元的數量。小鼠灌注取腦,制備20 μm厚切片,每只小鼠留腦片4張。腦片以40 g/L多聚甲醛溶液固定10 min,應用0.01 mol/L PBS溶液洗3次(每次10 min),山羊血清封閉2 h,加TH一抗孵育過夜;用0.01 mol/L PBS 溶液洗3次(每次10 min),加二抗孵育;以0.01 mol/L PBS溶液洗3次(每次10 min),DAB顯色,中性樹膠封片。在20倍物鏡下計數每張腦片黑質區陽性神經元,將每張腦片中的陽性神經元總數再乘以 4,即得到黑質區 TH陽性神經元總數。
1.4 統計學處理
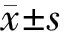
2 結 果
對照組、DFO組、LPS組和LPS+DFO組黑質區TH陽性神經元總數分別為(14.75±1.75)×103、(12.14±2.32)×103、(5.26±1.42)×103和(11.14±1.18)×103(n=6)。析因設計的方差分析顯示,DFO和LPS兩種因素存在交互作用(F=36.40,P<0.05)。主效應分析顯示,LPS組與對照組相比,TH陽性神經元總數明顯降低,差異有統計學意義(F=55.58,P<0.05);DFO組與對照組相比,TH陽性神經元總數降低,差異亦有統計學意義(F=5.58,P<0.05)。單獨效應分析顯示,DFO組與對照組相比,TH陽性神經元總數差異無顯著性(F=4.84,P=0.05);LPS組與對照組相比較,TH陽性神經元總數差異有統計學意義(F=106.15,P<0.05);LPS+DFO組與LPS組相比較,TH陽性神經元總數差異有統計學意義(F=60.74,P<0.05);而LPS+DFO組與DFO組相比,TH陽性神經元總數差異無顯著性(F=0.88,P=0.37)。
3 討 論
PD作為僅次于阿爾茨海默病的第二大神經退行性疾病,其主要病理特征是中腦黑質多巴胺能神經元的進行性喪失和紋狀體多巴胺耗竭[8],與小膠質細胞誘導的神經炎癥、氧化應激及線粒體功能障礙密切相關[9-10]。大量研究結果表明,神經炎癥在PD的發病過程中起著關鍵作用,源自非神經元細胞(包括小膠質細胞)的炎癥遞質,如活性氧(ROS)、一氧化氮、腫瘤壞死因子α(TNF-α)和白細胞介素1β(IL-1β)均可以調節PD中神經元細胞死亡的進程[11-13]。紋狀體內注射LPS可激活小膠質細胞,引起炎癥反應,導致羥自由基的產生及細胞外多巴胺、谷氨酸和腺苷水平上升,并引發多巴胺能神經元損傷反應[14]。
鐵作為中樞神經系統生長及發育所必需的微量元素,在正常衰老和神經退行性疾病的許多生物過程中起著重要作用,不僅參與調節線粒體產生能量,還與中樞神經系統的正常髓鞘形成密切相關[15]。大量研究顯示,PD病人存在鐵代謝異常,病人腦中鐵逐漸積累,特別是在黑質致密部[16-17]。鐵介導的多巴胺能神經元損傷與其參與ROS的生成有關,過氧化氫在多巴胺氧化脫氨基過程中產生,它與Fe2+反應,產生羥基自由基,破壞蛋白質、核酸和膜磷脂等,進而引起多巴胺能神經元損傷[18]。
鐵螯合劑作為PD的潛在治療藥物,通過降低不穩定的鐵的水平來預防和治療模型小鼠PD[15]。DFO可以螯合過多的三價鐵,清除過量游離鐵,抑制羥自由基生成,從而抑制氧化應激反應,起到保護多巴胺能神經元的作用。有研究證實,DFO預處理可以改善LPS誘導的小膠質細胞激活和降低海馬中升高的IL-1β和TNF-α水平,阻止LPS誘導的海馬中丙二醛和超氧化物歧化酶水平的增加,并降低其誘導的鐵積累[7]。本實驗室前期研究證實,神經炎癥會激活小膠質細胞,通過白細胞介素6信號,刺激星形膠質細胞釋放鐵調素,向神經元發送信號,調節神經元鐵代謝相關蛋白的表達,導致神經元鐵的沉積,進而引起神經元損傷[19-21]。本實驗結果顯示,與LPS組相比,DFO聯合LPS處理組黑質TH陽性多巴胺能神經元數量明顯增多,這可能與DFO螯合鐵從而降低ROS水平有關。
總之,本實驗結果表明,鐵螯合劑DFO具有防止LPS誘導的多巴胺能神經元死亡的神經保護作用。在此基礎上,我們將繼續探索其相關機制,為對抗神經炎癥的生物學效應開辟一條新的途徑,也為PD的治療提供新的實驗和理論基礎。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
東方少年·布老虎畫刊(2023年8期)2023-08-01 15:45:12
科學大眾(2021年6期)2021-07-20 07:42:44
科學(2020年3期)2020-11-26 08:18:30
學苑創造·A版(2020年9期)2020-10-13 09:41:02
娃娃樂園·綜合智能(2019年3期)2019-04-03 09:17:36
中成藥(2018年2期)2018-05-09 07:19:34
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
湖北師范大學學報(自然科學版)(2015年2期)2016-01-10 08:41:55
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
