數據挖掘探究肝-腎-肺纖維化的關鍵基因
2022-12-17 03:21:26閆慧芳
生物信息學 2022年4期
閆慧芳
(中國人民大學 統計學院,北京 100872)
器官纖維化是細胞外基質(Extracellular Matrix, ECM)的過度積累,從而導致組織結構的扭曲及器官功能的喪失。很多慢性病可引起纖維化,包括糖尿病、高血壓、病毒性、慢性肝病等疾病導致的纖維化可因過量的ECM取代和破壞實質組織而致肝、肺、腎、心臟或其他重要器官的衰竭[1]。最近的分析表明,在西方發達國家,接近一半的死亡都與纖維性疾病有關[2]。解決器官纖維化問題的能力可能因為一些因素而不同,包括器官、有害刺激因素的性質以及患者的特征(例如年齡和遺傳背景)[3-5]。然而,來自人類和動物模型的很多研究表明,纖維化在所研究的大多數器官或組織中存在被逆轉的可能。
計算生物學方法在確定人類疾病的潛在遺傳基礎方面起著至關重要的作用[6]。高通量平臺例如微陣列技術和RNA測序技術可用于分析轉錄層面上基因的改變,越來越被視為在疾病研究中提供重要幫助的工具[7-9]。近年來,結合生物信息學分析的微陣列技術能夠快速處理大量的數據集,使人們能夠全面識別數百個與某些疾病的發生和發展有關的差異表達基因(Differential Expressed Genes,DEGs)。
針對器官纖維化也有許多研究工作,它們從不同的角度闡釋了不同組織間纖維化的共同特征。雖然組織成纖維細胞通常是異質性的,但病理性肌成纖維細胞在肺、腎或肝纖維化中表現出類似的組織學表型和分子特征,這表明基本的致病途徑在不同纖維化器官存在共性[10-12]。Zhu Z.等整合了3個微陣列公共數據集,利用生物信息分析識別了肝纖維化相關的基因和通路,此外還對潛在的治療藥物進行了初步篩選[13]。Chau-Chyun S.等針對特發性肺纖維化利用二代測序(Next Generation Sequencing, NGS)平臺獲取纖維化肺和健康肺的mRNA數據,利用逐步的生物信息學手段分析獲得與之相關的差異基因,他們也利用了Gene Expression Omnibus (GEO)公共數據集進行了驗證[14]。Wang S.等識別出CXCL14 是一個由不同疾病引發的肝纖維化的共有差異基因,它可能會成為治療多因素肝纖維化的潛在靶點[15]。Mira P.等利用實驗收集了腎纖維化相關的多組學數據(包括DNA,miRNA和蛋白),利用計算生物學方法進行了整合分析,并將這些數據和分析結果生成了在線工具,以供研究人員查找腎纖維化相關的轉錄組和蛋白組信息[16]。Zhao J.Q.等對纖維化的心血管疾病進行了差異基因分析和功能富集,最終識別出11個關鍵基因[17]。Eugene M.等利用計算方法分析了肝纖維化和肺纖維化的共有代謝通路,為探究纖維化治療方法提供了新思路[18]。雖然近年來研究人員已經利用生物信息學對纖維化進行了一系列研究,仍有以下幾方面需要進一步探究:1)不同器官纖維化的共有關鍵基因尚不完全明確;2)肝纖維化的特有關鍵基因信息鮮有研究;3)此外,為了發現潛在藥物靶點,針對關鍵基因及蛋白的成藥性分析是十分必要的[19]。
本文利用計算生物學方法對GEO公共數據集 (GSE36066, GSE97546, GSE55747) 進行了整合分析和數據挖掘。其中GSE36066和 GSE97546包含 肝、腎、肺各一組數據,先基于它們進行了第一輪分析。之后另外引入一個肝纖維化數據集GSE55747做進一步的驗證。最后針對驗證后的纖維化關鍵基因,進行了成藥性的評估。這些研究將有助于進一步理解纖維化過程的內在機理,也可為纖維化生物標志物和潛在藥物靶點的發現提供借鑒。
1 材料與方法
1.1 數據集
從GEO數據庫中下載3個基于小鼠的mRNA數據集,其中GSE36066和GSE97546用于差異表達基因和相關功能富集等的分析,GSE55747用于結果的驗證。3個數據集的基本信息(見表1)。其中GSE36066為膽管結扎誘發的肝纖維化樣本與非纖維化樣本,GSE97546包含單側輸尿管梗阻誘發的腎纖維化和博來霉素誘發的肺纖維化樣本及相應的對照樣本,驗證數據集GSE55747則是四氯化碳誘發的肝纖維化樣本與非纖維化肝樣本。3個數據集的采集基于不同的微陣列平臺,數據樣本的對照組和處理組樣本均有生物學重復。

表1 數據集的基本信息Table 1 Basic information of datasets
1.2 差異表達基因(Differentially Expressed Genes, DEG)的分析及選擇
GEO2R(http://www.ncbi.nlm.nih.gov/geo/geo2r)提供了友好用戶界面,能夠對GEO數據進行基于R的統計分析。 GEO2R是一種智能的在線分析工具,可以在相同的實驗條件下既能整合分析兩個數據集,也可以拆分任何GEO數據[20]。本研究應用GEO2R工具分析纖維化器官與正常器官之間差異表達基因。調整p值 < 0.01,log2FC(Fold Change)的絕對值 > 1.0的基因被認為是差異表達基因DEGs。獲取差異表達基因后利用在線Venn分析工具(http://bioinformatics.psb.ugent.be/webtools/Venn)獲得肝、腎、肺纖維化的DEG交集及肝纖維化特有DEG。
1.3 GO(Gene Ontology)功能富集及KEGG信號通路分析
基因本體論(Gene Ontology, GO)是一種常用的生物信息學工具,可根據定義的特征提供有關單個基因組產品的基因功能的全面信息。GO富集分析能從三方面更好地解釋基因的功能:分子功能(Molecular Function, MF),生物過程(Biological Process, BP)和細胞成分(Cellular Component, CC)。KEGG(Kyoto Encyclopedia of Genes and Genomes, KEGG)是系統分析基因產物在細胞中的代謝途徑以及這些基因產物功能的數據庫。本研究中,使用Webgestalt(http://www.webgestalt.org)進行了GO和KEGG分析[21]。
1.4 驗證分析
利用數據集GSE55747對以上分析獲取的差異基因進行驗證。首先使用GEO2R在線平臺分析獲得該數據集中肝纖維化樣本與對照樣本的差異表達基因。調整p值<0.01,log2FC(Fold Change)的絕對值>1.0的基因被認為是差異表達基因DEGs。然后利用Venn分析工具對GSE55747的DEGs與先前識別出的三種器官纖維化共有差異基因和肝纖維化特有差異基因進行比較分析,取兩者的交集,從而得到進一步驗證的差異表達基因列表。
1.5 蛋白質-蛋白質相互作用 (Protein-Protein Interaction, PPI)及關鍵基因識別
STRING(Search Tool for the Retrieval of Interacting Genes , STRING)[22]數據庫提供了實驗和預測的相互作用信息。在本研究中,STRING在線工具用于分析DEG綜合得分 > 0.4的PPI。然后通過Cytoscape可視化PPI網絡[23],利用其中的CytoHubba插件[24]分別計算得到3種器官纖維化共差異基因和肝纖維化特有差異基因PPI網絡中前10個具有最高連接度的基因(蛋白),將其視為關鍵基因(蛋白)。
1.6 關鍵基因/蛋白的成藥性(Druggability)評估
本研究主要基于3個維度的信息評估關鍵基因/蛋白的成藥性。以往的研究表明藥物-蛋白相互作用的發生區域對于藥物的合理遞送是十分重要的[25],因此我們將基因/蛋白的分布區域作為評估的第1個維度,如果蛋白分布于胞外或細胞膜,則賦分2分,其他分布區域賦分為0分。通常分泌蛋白的性質會利于該蛋白的成藥性[26]。因此是否有證據表明蛋白為分泌蛋白,是評估的第2個維度,分泌蛋白賦分1分,非分泌蛋白賦分0分。此外有些蛋白并沒有直接的證據表明它們分布于胞外或為分泌蛋白,但是曾有研究在血漿中檢測到它們的存在,這一信息也是成藥性的因素之一。本研究將血漿中曾檢測到濃度的蛋白賦分1分,其余賦分0分。最后對各關鍵蛋白/基因的三項賦分加和得到最終的成藥性評分,0-4分代表成藥性由低到高。所獲取的蛋白分布、是否為分泌蛋白等信息均來自于HPA(The Human Protein Atlas) 數據庫[27]。
2 結果分析
2.1 多器官纖維化共有差異表達基因(DEGs)及肝纖維化特有差異表達基因
利用GSE36066和GSE97546數據集,識別了肝、腎、肺三種器官纖維化的各自的差異表達基因。adj.p值 < 0.01,log2FC(Fold Change) > 1.0作為界定差異基因的標準。通過GEO2R分析,GSE36066肝纖維化DEG 2 382個,GSE97546腎纖維化 DEG 2 632個,GSE97546肺纖維化DEG 2 897個 (見圖1)。隨后對三種器官纖維化的DEG進行韋恩分析,得到多器官纖維化的共有DEGs 196個和肝纖維化特有DEGs 1 562個(見圖2)。
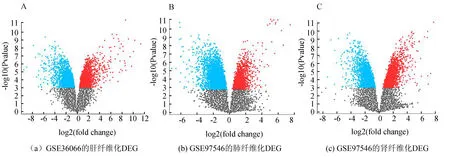
圖1 差異表達基因的火山圖Fig. 1 Volcano diagram of DEGs

圖2 差異表達基因的韋恩圖Fig. 2 Venn diagram of DEGs
2.2 GO功能富集和KEGG通路
為了進一步理解差異表達基因參與的生物過程和信號通路,利用Webgestalt在線工具對多器官纖維化共有DEG進行了GO功能富集和KEGG信號通路分析,另外對肝纖維化DEG進行了生物過程的功能富集分析。
2.2.1 三種器官纖維化共有差異基因的功能富集和信號通路分析
對肝、肺、腎三種器官纖維化共有的196個差異基因進行的GO功能富集和KEGG信號通路分析結果(見圖3)。采用有向無環圖(DAG)展示GO功能富集的分析結果,其中生物過程(BP)富集最顯著的包括炎癥反應、免疫反應、免疫系統調節等(見圖3a)。分子功能(MF)富集顯著的為脂磷酸結合、趨化因子活性、信號受體結合等(見圖3c)。而細胞組成(CC)分析顯示,這些基因表達的蛋白大部分為細胞外區域、細胞表面或細胞外壁的組分,這也一定程度上為蛋白/基因的成藥性提供了證據(見圖3b)。此外,KEGG通路分析的火山圖(見圖3d) 顯示差異表達基因在TNF信號,Toll受體信號、流體剪切應力與動脈粥樣硬化等通路中有較顯著的富集。

圖3 GO功能富集和KEGG信號通路分析Fig. 3 GO functional enrichment and KEGG signaling pathway analyses
2.2.2 肝纖維化特有差異基因的生物過程(BP)功能富集
對肝纖維化特有的1 562個差異基因進行的生物過程(BP)富集分析結果(見圖4)。BP富集分析中滿足p值和錯誤發現率(False Discovery Rate, FDR)均 < 0.05的生物過程被視為顯著,然后按照富集比率(Enrichment Ratio, ER)由大到小排列取前10項。結果表明這些差異基因的生物功能主要集中在脂肪酸代謝、羧酸代謝、脂質代謝、氧化還原等過程。

圖4 肝纖維化特有差異基因的生物過程富集Fig.4 Biological process enrichment for liver fibrosis specific DEGs
2.3 數據驗證獲得最終的差異表達基因
本研究除了探究三種器官纖維化共有關鍵基因以外,還研究了肝纖維化的特有關鍵基因,故引入一個肝纖維化數據集GSE55747做進一步的驗證。利用GEO2R對數據集GSE55747進行差異基因分析,同時滿足adj.pvalue<0.01 和log2FC>1.0被認為是顯著的差異表達基因DEG。然后利用Venn分析工具與先前得到的肝腎肺三器官纖維化共有差異基因(196個)和肝纖維化特有差異基因(1 562個)分別取交集。最終得到驗證后的肝-腎-肺三器官纖維化共有差異基因58個,肝纖維化特有差異基因85個(見表2)。

表2 驗證后的三器官纖維化共有差異基因和肝纖維化特有差異基因Table 2 Common DEGs of fibrosis in three organs and liver fibrosis specific DEGs after validation
2.4 蛋白質-蛋白質相互作用(PPI)及關鍵基因
為了更好地理解識別得到的共有差異基因和特有差異基因中哪些起到關鍵作用,利用STRING工具進行了蛋白質-蛋白質相互作用的分析,得到了包含58個nodes、145個edges的共有差異基因網絡和包含82個nodes、53個edges的肝纖維化特有差異基因網絡(見圖5)。然后利用Cotoscape中Cytohubba計算插件分別對這2個PPI網絡得到基于MCC (Maximal Clique Centrality) 法排序前10的基因。本研究中將這些基因作為與纖維化過程相關的肝腎肺共有關鍵基因和肝特有關鍵基因(見表3)。將這20個小鼠基因與人類基因進行匹配,得到14個匹配成功的對應人類基因。
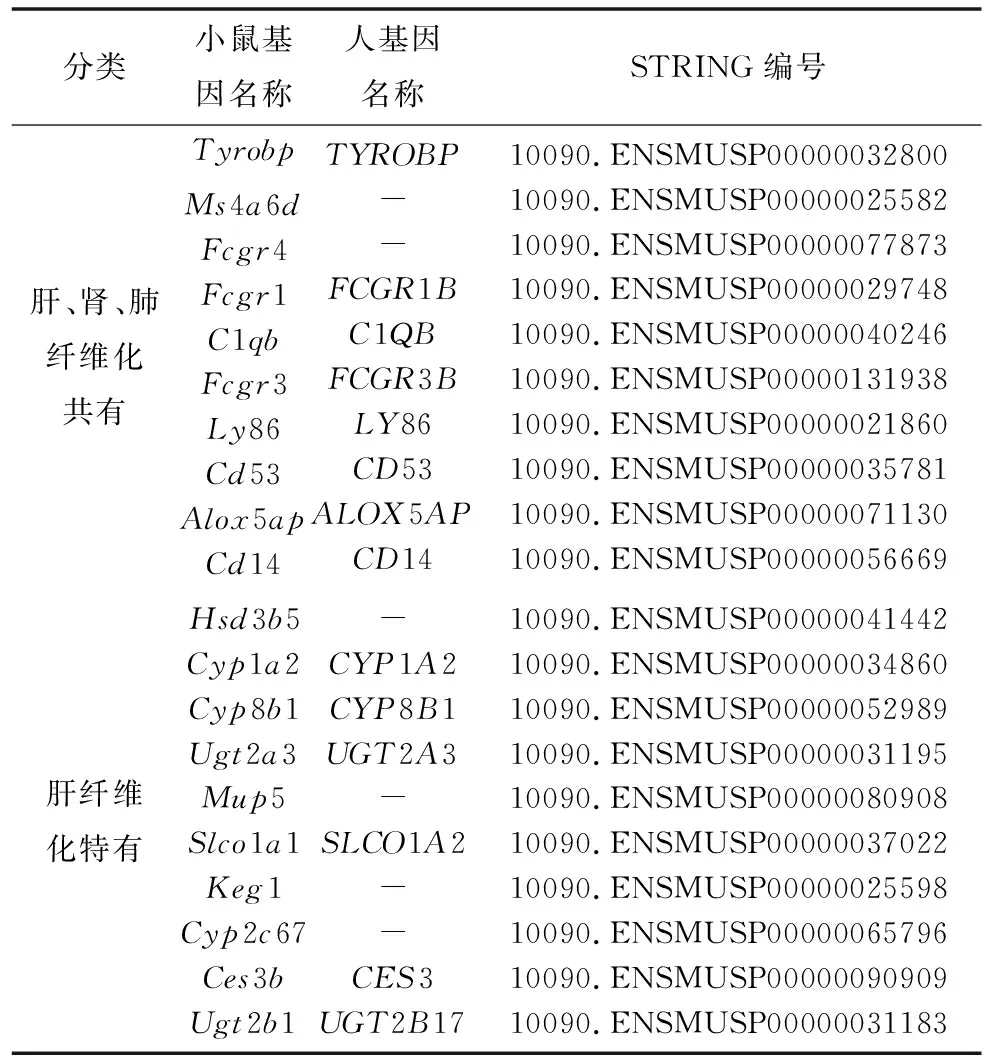
表3 多器官纖維化和肝纖維化特有的關鍵基因 (各取前10個)Table 3 Hub genes for multi-organ fibrosis and liver fibrosis only (ten for each)

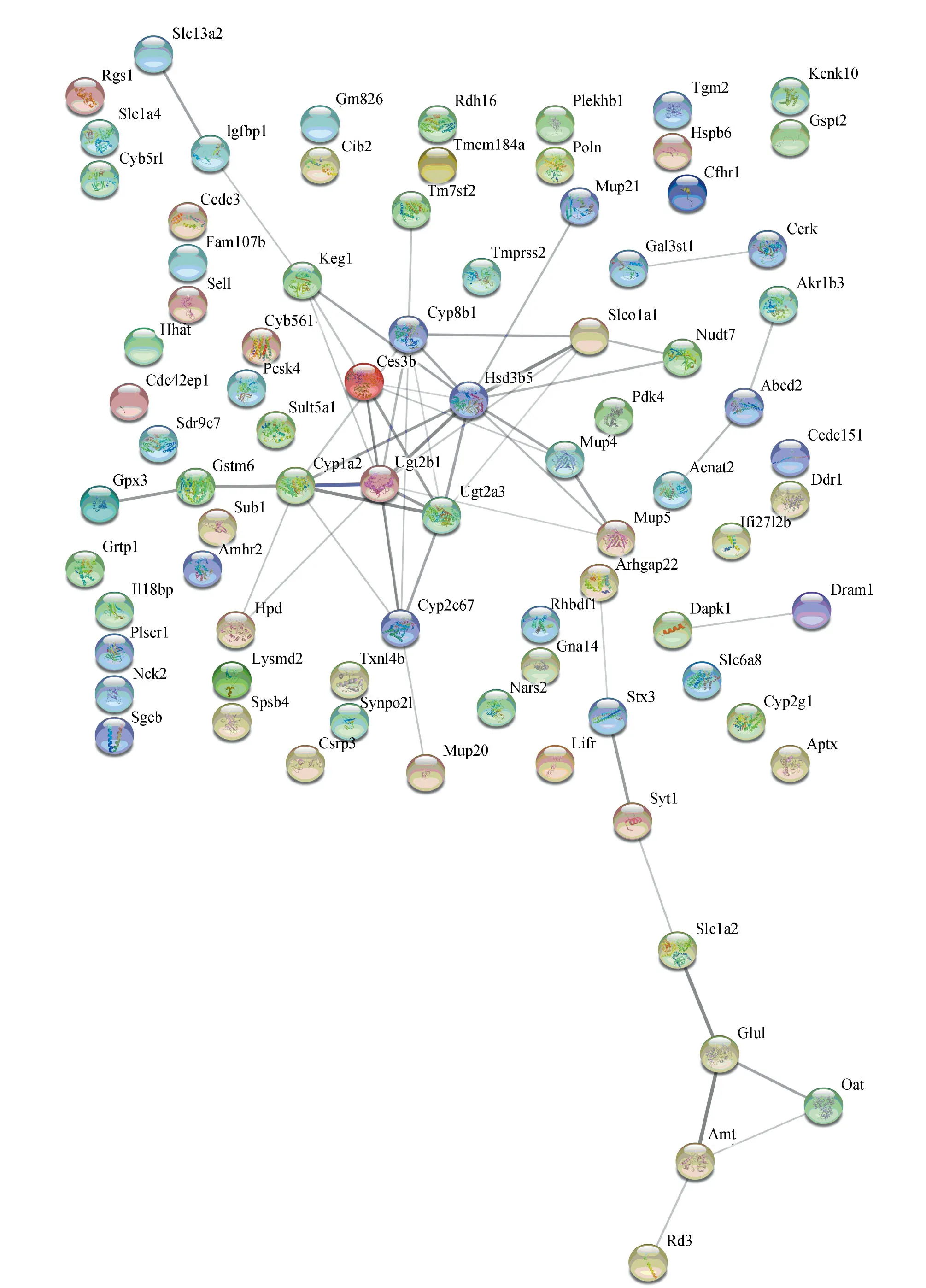
圖5 蛋白質-蛋白質相互作用網絡Fig.5 Protein-protein interaction network
2.5 關鍵基因的成藥性
蛋白分布區域、是否為分泌蛋白以及是否有研究在血漿中檢測到為3個考慮的關鍵指標,這些指標對應的信息全部來自于公共數據庫。針對這些信息,進行相應的賦分,其中分布區域為成藥性最重要的因素,因此分布于胞外或細胞膜則賦2分。如果為分泌蛋白或有血漿中濃度信息各賦1分。將這三項的賦分加和,即得到最終的成藥性評分。經過分析計算,在這14個關鍵基因中,有3個具有較高的成藥性(4分),6個有中等的成藥性(2~3分), 5個具有較弱的成藥性(0~1分),具體結果(見表4)。
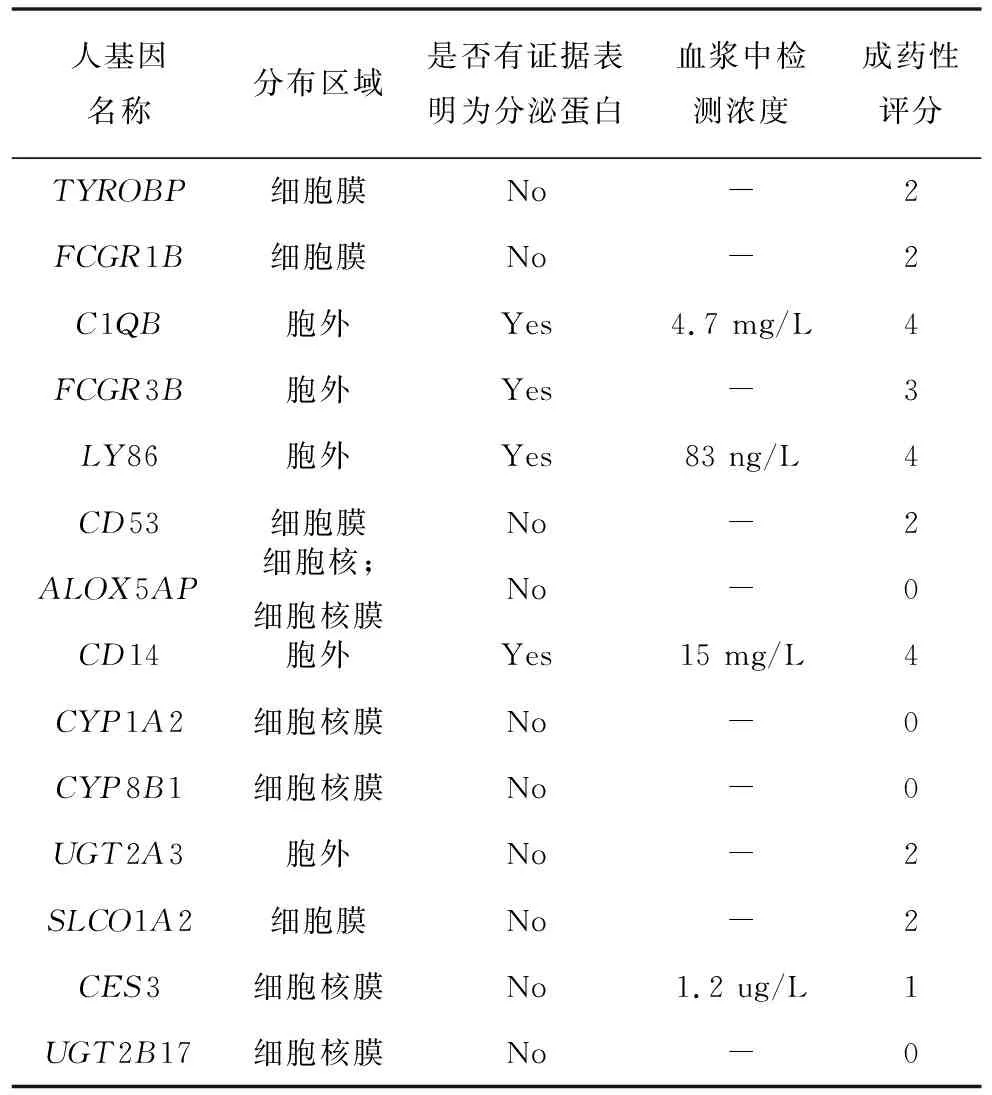
表4 關鍵基因的成藥性信息Table 4 Druggability information for hub genes
3 討 論
纖維化是細胞外基質的過度積聚,通常是由于重復或慢性組織損傷的傷口愈合反應,并可能導致器官結構破壞和功能的喪失。雖然纖維化在以前被認為不可逆,最新的證據表明某些情況下纖維化疾病是可以被逆轉的,纖維化消退的機制包括降解纖維化的細胞外基質以及消除相關的成纖維細胞。已有研究表明不同器官的纖維化機制存在共性,因此為了探究與纖維化過程相關的關鍵基因和信號通路,本研究通過對公共數據集的數據挖掘,識別了肝臟、腎臟及肺臟三種器官纖維化的共有關鍵基因和相關生物通路。此外,也對肝纖維化的特有差異基因進行了分析。研究人員做了大量基于動物模型的纖維化研究,雖然動物模型并不能完全描述相應的人類疾病,但是相比于人的試驗,動物試驗具有更加可控可比的樣本組和對照組,也利于采集噪音更小的數據。已經有許多具有代表性的纖維化動物模型。其中膽總管結扎和四氯化碳誘導作為代表性的肝纖維化動物模型構建方法。膽管結扎動物模型在全世界數百個實驗室中用于誘發肝膽汁淤積和纖維化。它誘導肝內膽管上皮細胞增生,使增殖的膽管上皮細胞周圍的門脈成纖維細胞發生肌纖維母細胞分化,從而導致ECM的高度復制,大量表達和沉積。因此,該模型在大鼠和小鼠中的應用在研究肝炎癥和纖維化發病機理的十分流行。四氯化碳誘導的動物模型主要機制為四氯化碳在肝臟中被細胞色素P450代謝,轉化為高反應性三氯甲基(CCl3)自由基,最終導致肝毒性損害、炎癥和纖維化。CCl4誘導的肝損傷構成了人類病理學的可靠模型,在生理和細胞水平上都模仿了其關鍵特征。對于腎纖維化,單側輸尿管梗阻(UUO)會在阻塞的腎臟中引發一系列事件,導致腎血流量和腎小球濾過率在24 h內急速下降。隨后的間質反應包括間質炎癥,腎小管擴張和腎小管凋亡,并從7 d開始導致腎小管萎縮和纖維化。盡管完全的輸尿管梗阻不是人類腎臟疾病的最常見原因,但由于UUO具有產生進行性腎纖維化的能力,它已成為慢性腎臟疾病的最受歡迎模型之一。完整的梗阻模型的優點是可重復性好(因此動物間的差異不成問題),時程短,性能容易,并且有對側腎臟作為對照。此外,該模型在大鼠和小鼠中都很容易誘導。而博來霉素誘導的動物模型是最具代表性的肺纖維化實驗模型。博來霉素是一種治療用的抗生素。當淋巴瘤患者靜脈注射BLM后出現肺纖維化時,已被確定為促纖維化藥物。它已被用于多種物種的模型構建,包括小鼠,大鼠,豚鼠,倉鼠,狗和靈長類動物。本研究中選用基于以上動物模型收集的數據集,利用GSE36066(肝纖維化)和GSE97546(腎纖維化和肺纖維化)識別出肝-腎-肺纖維化共有差異表達基因196個,肝纖維化特有差異表達基因1 562個。然后引入數據集GSE55747進行了驗證分析,得到驗證后的共有DEG 58個,肝纖維化特有DEG 85個。肝-腎-肺纖維化共有DEG主要富集于炎癥反應(Inflammatory Response)和免疫反應(Immune Response)過程。以往的很多研究表明,化學物質、微生物或生理組織損傷引起的慢性炎癥通常會導致纖維化病變[28-30],本研究的富集結果與這一結論相吻合。尚未有研究表明存在未發生炎癥反應而產生的纖維化,而不同原因和不同器官中發生的纖維化通路可能十分相似[31]。生物過程(BP)富集結果顯示196 個肝腎肺共有DEG中約50個被歸類于免疫反應或炎癥反應過程。其中CCL5、CCL6、CCL12、CXCL1、CXCL14和CX3CL1屬于趨化因子(Chemokines)超家族,趨化因子是一類參與免疫調節和炎癥過程的蛋白[32]。研究表明趨化因子參與所有的傷口愈合反應且在纖維化過程中起到通用的及器官特異性的作用[33]。CD14、CD44和CD84基因編碼的蛋白都屬于表面抗體,優先在巨噬細胞和白細胞上表達。CD14與其他蛋白質協同介導免疫應答[34]。CD44編碼的蛋白質是一種細胞表面糖蛋白,參與細胞間相互作用,細胞粘附和遷移。已被證實CD44與其配體HA與許多炎癥性疾病有關[35]。CD84編碼一種膜糖蛋白,該蛋白是信號淋巴細胞激活分子(SLAM)家族的成員。研究顯示CD84介導的信號調節多種免疫過程[36]。C1qa和C1qb分別編碼血清補體亞成分C1q的A鏈和B鏈多肽,而C1q與自身免疫和細胞凋亡密切相關[37]。
肝纖維化特有DEG的富集分析顯示與脂肪酸代謝(Fatty acid metabolic process)和脂質代謝(Lipid metabolic process)過程有很強的聯系。富集分析表明1 562 個肝纖維化特有DEG中約160個被歸類于脂質代謝或脂肪酸代謝過程。肝臟為脂質代謝的重要器官,而數據集GSE36066和GSE55747中肝纖維化小鼠模型分別基于膽管結扎(BDL)和四氯化碳(CCL4)誘導。以往的研究表明,BDL誘導會降低肝臟中細胞色素P450含量、過氧化脂肪酸的beta-氧化和催化活性,而這些變化并不會發生在腎臟中[38]。也有很多研究表明CCl4會引發脂質過氧化,從而造成肝損傷[39-42]。因此相較于腎臟和肺臟纖維化,在肝纖維化中特異性地識別出脂質代謝相關的差異基因是合理的。而這些與脂質代謝或脂肪酸代謝相關的基因是否特異性地在肝纖維化中發揮作用,還需要進一步的驗證與分析。
利用PPI分析得到肝-腎-肺纖維化共有和肝纖維化特有關鍵基因各10個,經過小鼠基因與人基因的匹配,成功識別出肝-腎-肺纖維化共有的人關鍵基因8個,肝纖維化特有的人關鍵基因6個。通過文獻調研發現,此前的研究有證據表明這些關鍵基因中5個與纖維化相關(TYROBP,FCGR3B,ALOX5AP,CD14,CYP1A2),3個與肝病相關(CYP8B1,UGT2A3,CES3)。TYROBP(DAP12)編碼包含基于免疫受體酪氨酸激活模體的跨膜信號多肽,參與淋巴與非淋巴細胞免疫調節的相互作用。Zhang L.等的研究表明在原發性膽汁性肝硬化(PBC)中TYROBP基因下調[43]。Tammaro A.等則發現TYROBP(DAP12) 參與了UUO誘發的腎纖維化[44]。FCGR3B編碼的蛋白是針對γ免疫球蛋白(IgG)Fc區的低親和力受體。它能夠充當單體,與單體或聚集的IgG相結合,可能在捕獲外周循環中免疫復合物過程中起到一定作用。Bournazos S等在一項病例對照研究中,使用CD36作為基因復制對照,通過實時定量PCR比較了142例IPF患者和221例對照中FCGR3B的拷貝數。證明了該基因拷貝數的差異(Copy Number Variation, CNV)與特發性肺纖維化有關[45]。ALOX5AP編碼一種是白三烯合成所必需的蛋白,白三烯是花生四烯酸代謝產物,已經證實與哮喘、關節炎等多種炎癥反應有關。針對囊性纖維化疾病,靶向ALOX5AP的藥物Fiboflapon在臨床二期研究中。另外Kowal-Bielecka O等的研究表明ALOX5AP的遺傳變異可能在肺纖維化中起作用[46]。CD14編碼的蛋白屬于表面抗體,優先在巨噬細胞和白細胞上表達,CD14與其他蛋白質協同介導免疫應答反應。Zhao SX等人的研究表明CD14陽性的單核細胞與 CD163陽性的巨噬細胞慢性丙型肝炎患者肝纖維化的嚴重程度相關[47]。Fukushima H的研究則表明,在具有纖維化非酒精性脂肪肝炎(NASH)的動物模型中,CD14陽性的肝巨噬細胞數量增加[48]。CYP1A2編碼一種細胞色素P450酶, 細胞色素P450是單加氧酶,可催化涉及藥物代謝和膽固醇和其他脂質合成的許多反應。Wuensch T等在非腫瘤肝組織中,觀察到CYP1A2活性逐漸下降,并伴有纖維化加劇[49]。在肝特異相關的關鍵蛋白中,CYP8B1在此前的研究中被證明與非酒精性脂肪肝病NAFLD(Non-alcoholic Fatty Liver Disease)相關。 Raphael C等通過研究發現在膽固醇誘導的NAFLD模型中,敲低Cyp8b顯著降低了脂肪變性和肝脂質含量[50]。Hardwick RN 等發現UGT2A3在非脂肪非硬化的非酒精性脂肪肝炎NASH病人中,表達水平增高,而在脂肪性NASH和肝硬化病人中并未發現這種差異[51]。Quiroga AD在大鼠早期肝癌中發現了CES3基因的下調,證明該基因可能與肝癌的發生發展有關[52]。
研究疾病的機制和關鍵基因的主要目的是開發更有效的藥物或治療手段。而這些關鍵基因作為潛在藥物靶點的一個重要前提是較高的成藥性 (Druggability)。基于蛋白分布區域、是否為分泌蛋白以及檢測到的血漿濃度進行的成藥性的分析顯示,7個共有關鍵蛋白/基因和2個特有關鍵蛋白/基因具有較高的成藥性。綜合富集結果、蛋白-蛋白相互作用、研究證據和成藥性信息,結果顯示TYROBP, FCGR3B, ALOX5AP和CD14或可作為纖維化治療的潛在靶點, 而CYP8B1和UGT2A3可能成為NAFLD或NASH等肝病治療的研究重點。對于本研究中識別出的關鍵基因FCGR1B, C1QB, LY86和CD53,雖然此前未有研究表明與纖維化直接相關,也可作為全新的靶點進行更深入的探究和驗證。
綜上所述,本研究利用計算生物學方法對GEO公共數據集進行了整合分析和數據挖掘,識別了肝臟、腎臟及肺臟三種器官纖維化的共有關鍵基因和肝纖維化的特有關鍵基因。GO功能富集和KEGG通路分析表明,肝-腎-肺纖維化共有DEG主要富集于炎癥反應和免疫反應過程,而肝纖維化特有DEG脂肪酸代謝和脂質代謝過程有很強的聯系。最后還對這些關鍵基因進行了成藥性的評估。綜合富集結果、蛋白-蛋白相互作用、研究證據和成藥性信息給出了纖維化和肝病的潛在藥物靶點。這些研究將有助于理解纖維化過程的內在機理,也為纖維化疾病的治療提供更多的可能。本研究全部基于動物模型公共數據集的分析和挖掘,并未經過實驗結果的驗證,未來通過體外和體內試驗對這些基因進行更深入的研究將有助于進一步明確它們在纖維化過程中發揮的作用。
4 結 論
1)TYROBP,FCGR3B,ALOX5AP和CD14或可成為纖維化治療的潛在靶點;
2)CYP8B1和UGT2A3可能與非酒精性脂肪性肝病(Non-alcoholic Fatty Liver Disease, NAFLD)或非酒精性脂肪性肝炎(Non-Alcoholic SteatoHepatitis , NASH)相關;
3)對于基因FCGR1B,C1QB,LY86和CD53,目前沒有直接的證據表明其與纖維化相關,需要進一步驗證它們在纖維化發生過程中的功能。
猜你喜歡
中老年保健(2022年2期)2022-11-25 23:46:31
昆明醫科大學學報(2022年4期)2022-05-23 13:04:50
中學生數理化(高中版.高考理化)(2021年6期)2021-07-28 06:21:04
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
國際呼吸雜志(2019年21期)2019-11-25 09:52:20
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
天津醫科大學學報(2015年3期)2015-06-05 12:21:49
NBA特刊(2014年7期)2014-04-29 00:44:03
中國商人(2013年1期)2013-12-04 08:52:52
