維格列汀的合成工藝研究
2022-12-09 12:27:16王麗霞劉新元王宇棟梁丙辰
煤炭與化工 2022年10期
關鍵詞:實驗
王麗霞,劉新元,王 鵬,王宇棟,梁丙辰
(1.河北合佳醫藥科技集團股份有限公司,河北 石家莊 052165;2.河北合佳醫藥科技集團 合佳研究院,河北 石家莊 050035)
0 引 言
維格列汀(Vildagliptin),化學名稱(S)-1-[[(3-羥基-1-金剛烷胺)氨基]乙酰基]-2-氰基吡咯烷,是瑞士諾華研發的一款二肽基肽酶抑制劑(DPP-4),2007年9月獲得歐盟批準上市。2011年8月,維格列汀正式獲得CFDA批準上市,商品名為“佳維樂”。本品適用于治療Ⅱ型糖尿病,可單獨使用也可與雙胍類、噻唑烷二酮類、磺脲類藥物聯合使用。其耐受性好,能有效控制血糖,且副作用少。原研廠家諾華公司的專利已于2019年到期,因此具有較大的應用價值和廣闊的市場前景。截至目前,國內外報道的維格列汀的合成路線大多都是以L-脯氨酰胺或L-脯氨酸作為起始原料,先合成關鍵中間體(S)-1-(2-氯乙酰基)吡咯烷-2甲腈,最后與中間體3-氨基-1-金剛烷醇進行縮合反應制備維格列汀。
如原研專利首先報道了以L-脯氨酰胺為起始原料,經取代反應和脫水反應得到關鍵中間體(S)-1-氯乙酰基-2-氰基吡咯烷,再與3-氨基-1-金剛烷醇反應得到目標產物維格列汀。該路線步驟較少,但所用原材料成本較高,且所得目標產物需通過柱層析提純,不利于工業化生產。
2004年的Bioorg Med Chem期刊報道了Fukushima H等的合成方法,以L-脯氨酸為原料,經二碳酸二叔丁酯保護氨基,再在1-(3-二甲胺基丙基)-3-乙基碳二亞胺(EDC)作用下與氨氣發生羰基氨基化反應,隨后用鹽酸脫去Boc保護基得L-脯氨酰胺,再繼續氯乙酰化、脫水、取代等一系列反應制得維格列汀。該方法原料來源廣泛,但工藝路線較長,整體收率偏低。
日本專利JP201356872報道了以(S)-吡咯烷-2甲腈鹽酸鹽為起始原料,在非均相體系中與氯乙酰氯制備關鍵中間體,工藝路線較短,但該起始原料極易吸潮,不便于制備、儲存、投料。
本文綜合上述方法的優點,以(S)-吡咯烷-2甲腈對甲苯磺酸鹽為起始原料,經氯乙酰化、親核取代兩步反應合成原料藥維格列汀。在其合成中,避免了催化劑碳酸鉀、碘化鉀的使用,且采用水作為溶劑,反應條件溫和易于控制。
1 實驗部分
1.1 主要試劑及儀器
1.1.1 試 劑
(S)-吡咯烷-2甲腈對甲苯磺酸鹽(石家莊合佳醫藥科技集團股份有限公司合佳研究院自制);氯乙酰氯(AR,羅恩試劑);三乙胺(AR,MREDA);3-氨基-1-金剛烷醇(AR,阿拉丁),其余試劑均為分析純。
1.1.2 儀 器
高效液相色譜儀(e2695,Waters)[面積歸一化法:色譜柱:依利特Hypersil ODS2(4.6×250 mm,5μm)流動相:乙腈/磷酸二氫鉀緩沖溶液(V/V=30∶70),檢測波長:210 nm,流速:1.0 mL/min,進樣量:10 μL,柱溫:35℃];紅外光譜儀(TensorⅡ,Bruker);熔點儀(IA9200,Electrothermal)。
1.2 實驗部分
本實驗的合成路線如圖1所示。
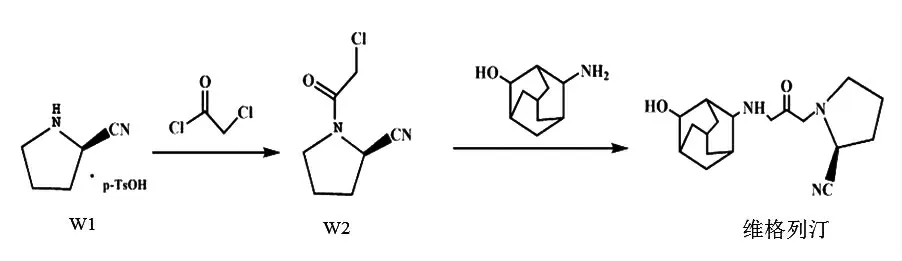
圖1 維格列汀合成路線Fig.1 Synthesis route of vildagliptin
1.2.1 W2的合成
在反應瓶中加入20 g(1eq)(S)-吡咯烷-2甲腈對甲苯磺酸鹽,8.75 g(1.2eq)三乙胺和160 mL二氯甲烷,冷卻至5℃左右,攪拌下滴加9.27g(1.1eq)氯乙酰氯,滴畢(15 min),繼續反應2 h。反應結束后,依次用飽和Na2CO3(30 mL)和飽和食鹽水(50 mL)洗滌反應液,無水硫酸鈉干燥,減壓蒸除溶劑,得11.72 g白色固體W2,收率91%,純度99.2%(HPLC),熔點64~66℃。IR(KBr,cm-1)v:2 953(-CH2),2 887(-CH2),2 241(-CN),1 661(-C=O),1 424(-CH2),1 345(-CH2),1 284(-C-N)。
1.2.2 維格列汀的合成與精制
向反應瓶中加入5 g(1eq)W2和9.67 g(2.0eq)3-胺基-1-金剛烷醇,25 mL水,升溫至45℃,攪拌下反應8 h后結束。加入30 ml甲苯洗滌2次;后降至室溫,并用10%鹽酸調節pH值至4.3,加入30 mL二氯甲烷洗滌2次;水相繼續加入100 mL二氯甲烷,溫度降至0~5℃,然后用Na2CO3溶液調節pH值至10.2,保留有機相;然后水相繼續用二氯甲烷(50 mL)進行萃取(50 mL×3),分液,合并有機相,蒸除溶劑;最后加入60 mL乙醇和乙酸乙酯(V/V=1∶4)進行精制,得維格列汀7.39 g,收率84.1%,HPLC純度99.9%(HPLC),熔點148~150℃。IR(KBr,cm-1)v:3 293(-NH,-OH),2 934(-CH2),2 849(-CH2),2 236(-CN),1 657(-C=O),1 405(-CH2),1 354(-CH2),1 252(-C-N)。
2 結果與討論
2.1 W2步單因素實驗
在W2的合成中主要考察了W1與氯乙酰氯的配比、W1與三乙胺的配比、反應溫度對產品純度、收率等的影響。因此預先按照上述操作進行試驗,之后保持其他因素不變,依次改變W1與氯乙酰氯、三乙胺的配比及反應溫度,考察各自對實驗的影響。
W1與氯乙酰氯的配比對實驗的影響見表1。
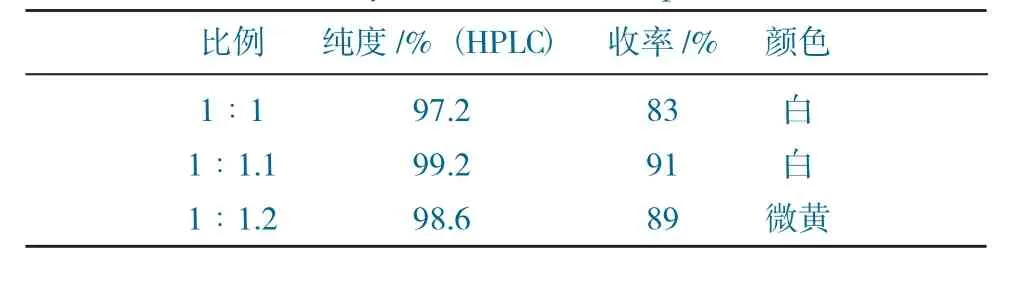
表1 W1與氯乙酰氯的配比對實驗的影響Table 1 The effect of the ratio of W1 to chloroacetyl chloride on the experiment
W1與三乙胺的配比對實驗的影響見表2。
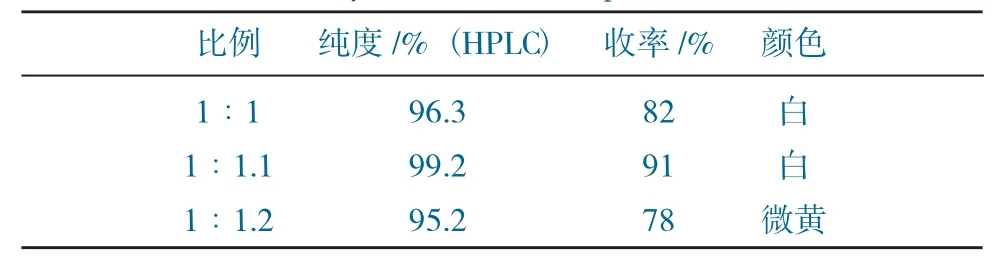
表2 W1與三乙胺的配比對實驗的影響Table 2 The effect of the ratio of W1 to triethylamine on the experiment
反應溫度對實驗的影響見表3。

表3 反應溫度對實驗的影響Table 3 The effect of the reaction temperature on the experiment
由表1~表3可得,當W1與氯乙酰氯的配比為1∶1時,純度及收率均不高,是氯乙酰氯量較少,導致W1原料有殘留,反應不完全,導致收率整體偏低;當配比提高為1∶1.2時,收率較高,但純度稍低,是過量的氯乙酰氯導致了雜質的生成,也影響了產品顏色。當W1與三乙胺的配比為1∶1.1時,純度及收率均不高,應是三乙胺未能將W1全部游離出來,導致反應不徹底;而將比例提高至1∶1.3時,產品顏色發黃,純度較低,可能由于三乙胺過量,后期水洗不徹底,導致產品發黃。當反應溫度逐漸升高,純度和收率均有不同程度降低,分析原因是由于溫度升高,導致體系中雜質的量增多,最終影響產品指標。
2.2 維格列汀單因素實驗
在維格列汀的合成中主要考察了W2與3-胺基-1-金剛烷醇的配比、反應溫度、反應時間對產品純度、收率等的影響。因此預先按照上述1.2(2)中操作進行試驗,之后保持其他因素不變,依次改變W2與3-胺基-1-金剛烷醇的配比、反應溫度及反應時間考察各自對實驗的影響。
W2與3-胺基-1-金剛烷醇的配比對實驗的影響見表4。
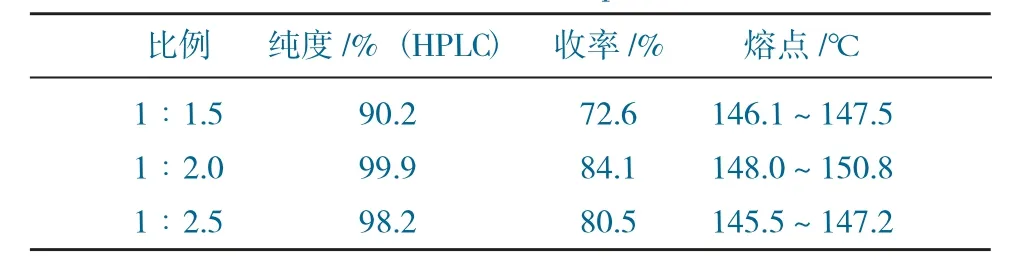
表4 W2與3-胺基-1-金剛烷醇的配比對實驗的影響Table 4 The effect of the ratio of W2 to 3-amino-1-amantadine on the experiment
反應溫度對實驗的影響見表5。

表5 反應溫度對實驗的影響Table 5 The effect of the reaction temperature on the experiment
反應時間對實驗的影響見表6。

表6 反應時間對實驗的影響Table 6 The effect of the reaction time on the experiment
由表4~表6可得,當W2與3-胺基-1-金剛烷醇的配比為1∶1.5時,純度較低,原料殘留較多,且維格列汀二聚體雜質較多,導致熔點 和收率均不高;將配比提高至1∶2.5時,收率尚可,但熔點偏低,可能是金剛烷醇較多,后處理未能除凈。
當反應溫度為室溫時,純度較低,反應較慢,48 h后原料仍有5%殘留;而當反應溫度提高至65℃時,反應速度較快,但收率和熔點偏低,可能由于高溫加速了維格列汀水解酰胺雜質等的生成,因此導致熔點、收率等受到影響。當反應時間為8 h時,原料殘留較多,導致收率偏低;但當延長反應時間至16 h時,體系中維格列汀酰胺雜質增多,加大了后處理難度,導致收率和熔點均偏低。
3 結 語
本研究以(S)-吡咯烷-2甲腈對甲苯磺酸鹽為起始原料,經兩步反應制得了原料藥維格列汀。同時還利用單因素實驗對各步實驗條件進行了優化,最終反應條件如2.1和2.2所述,最終純度達99.9%,總收率達76.5%。本研究采用的原料吸水性低、方便制備、儲存和投料,最后采用水作反應溶劑,降低了生產成本,減輕環境污染壓力,為維格列汀的工業化生產提供了參考。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
