急性胰腺炎肝損傷的分子機制
2022-11-16 08:42:26徐文倩吳瑤麒張近遠李合國
臨床肝膽病雜志 2022年11期
徐文倩,郭 敏,王 曉,吳瑤麒,張近遠,李合國
1 河南中醫藥大學 第一臨床醫學院,鄭州 450000;2 河南中醫藥大學第一附屬醫院 脾胃肝膽科,鄭州 450000
急性胰腺炎是各種刺激導致腺泡中胰酶被激活,造成各種炎性因子釋放而引起的由局部到全身的炎癥性反應。肝臟在急性胰腺炎過程中往往最易也最先受損,15%~60%的急性胰腺炎患者會合并肝損傷。急性胰腺炎作為一種消化系統急癥,1961年—2016年在歐美等國家發病人數每年增長率在2.77%~3.67%[1]。全球急性胰腺炎的發病率為33.74(95%CI:23.33~48.81)/100 000例,而其中每年1.60(95%CI:0.85~1.58)/100 000例死亡[2]。在死亡病例中因肝衰竭而死亡的人數占83%[3]。隨著飲食結構的改變,我國急性胰腺炎呈現年輕化、重癥化的特點[4]。隨著病情進展,肝臟在急性胰腺炎繼發多臟器損傷的過程中發揮著橋梁的作用。阻斷急性胰腺炎肝損傷有助于逆轉急性胰腺炎繼發多臟器損傷的進程。本文將從胰腺與肝臟的生理聯系、細胞因子、炎癥反應、氧化應激、微循環障礙、腸道菌群6個方面闡述急性胰腺炎肝損傷發生的分子機制,以期為防治急性胰腺炎并發多臟器損傷提供一定的參考。
1 胰腺與肝臟的生理聯系
在胚胎發育早期,胰腺起始于原腸尾部腹側和背側胰芽[5],這與肝臟發育起始幾乎位于同一區域,早期原腸外包裹著完整的膜系結構,伴隨著胚胎逐漸發育成熟,原腸外膜系結構發展成為腸系膜,近年來有假設提出這種膜系結構在肝臟和胰腺發育的過程中得以保留,成熟的肝、膽、胰等臟器通過這些系膜形成生理性粘連[6],由此推測胰腺與肝臟之間可能通過“系膜系統”的血管與淋巴管構成一個局部的微循環系統,并作為各種細胞因子的橋梁,在胰腺炎過程中對肝臟造成直接的損傷。從解剖學來看,肝臟通過門靜脈收集來自胰腺的血液。肝臟擁有雙重血供,使肝細胞生長于富氧環境中,這也決定了肝細胞較其他器官細胞對缺血缺氧環境更加敏感。而且肝和胰腺的淋巴結及神經纖維系統錯綜復雜,相互交織成網,這都為胰腺炎繼發肝損傷提供了支持。有研究[7]證實,切斷動物雙側內臟大神經能夠通過核因子-κB(NF-κB)途徑改善急性胰腺炎肝損傷程度。這在一定程度上反映了胰肝系膜系統的完整性。
2 細胞因子
人體細胞因子總量的50%在肝臟內產生,肝內Kupffer細胞作為人體最大的巨噬細胞群在細胞因子的釋放過程中發揮著重要的作用。肝臟內巨噬細胞受到胰腺釋放的損傷因子的刺激后,活化為具有介導炎癥反應的M1巨噬細胞和具有抗炎作用的M2巨噬細胞,M1和M2巨噬細胞在一定條件下可以相互轉化。近來有研究[8]證實,IL-4受體亞單位α通過激活ECM/Akt信號通路促使Kupffer細胞由M1向M2表型轉化,發揮抗炎作用,從而減輕肝細胞的損傷。M1巨噬細胞在向損傷部位募集的過程中能夠釋放IL-17、IL-1β、IL-6、IL-8、IL-23、IL-12、TNFα、巨噬細胞游走抑制因子(macrophage migration inhibitory factor, MIF)等多種細胞因子,誘導肝臟炎癥反應,引起肝細胞損傷。表1總結了急性胰腺炎過程中各種細胞因子對肝臟的作用。

表1 急性胰腺炎過程中細胞因子對肝臟的作用
3 炎癥反應
急性胰腺炎過程中所釋放的大量細胞因子是引起炎癥反應的物質基礎,急性胰腺炎初期是局部的無菌性炎癥,繼而引起全身各臟器的炎癥反應綜合征(systemic inflammatory response syndrome, SIRS),并伴隨補償性抗炎綜合征(compensatory anti-inflammatory response syndrome, CARS)。在急性胰腺炎肝損傷初期階段,JAK將STAT磷酸化,STAT二聚化后進入細胞核內進行相關基因表達,釋放TNFα、IL-18、IL-6等早期細胞因子[23]。與JAK釋放炎癥介質的途徑相仿,蛋白激酶C(protein kinase C, PKC)作為G蛋白偶聯系統的效應物,在外界刺激下發生磷酸化,繼而激活Iκ-B釋放基因調節蛋白NF-κB,NF-κB促使炎性細胞因子釋放,介導炎癥反應。TNFα能夠激活單核巨噬細胞、活化NF-κB,啟動巨噬細胞的吞噬作用,雖然該作用在炎癥初期有利于炎癥的消除,但在炎癥進展迅速的情況下會進一步加重肝臟炎癥反應,這些炎癥因子誘發了早期的炎癥反應(圖1)。TLR4作為一種病原識別受體,能夠識別最初胰腺釋放的胰酶與各種細胞因子信號,釋放炎癥反應的初始信號,啟動瀑布樣級聯炎癥反應。眾所周知,乳酸是人體細胞無氧代謝的產物,在體內過量堆積對人體造成不良影響,近年來有研究[24]發現,乳酸與特定受體結合后能夠顯著降低TNFα、IL-6等炎性因子的水平,有效減輕各種炎癥性肝病的肝損傷程度,在急性胰腺炎過程中,乳酸能夠與Kupffer細胞中的G-蛋白偶合受體81(Gi-protein-coupled receptor 81, GPR81)結合,降低NLRP-3炎性小體的表達,抑制半胱氨酸天冬氨酸轉移酶1(caspase-1)的活性,在一定程度上減輕急性胰腺炎大鼠的肝炎癥性損傷程度[25]。NLRP-3炎性小體是TLR4重要的下游信號分子,能夠介導caspase-1激活IL-1β、IL-18等炎癥因子,促進炎癥因子的成熟,引起肝臟的炎癥反應(圖1)。大量的動物實驗研究證實,NLRP-3炎性小體能夠通過加重急性胰腺炎過程中的炎癥反應誘導肝損傷以加重SIRS和CARS[26]。肝細胞高遷移率族蛋白1(hepatocyte high mobility group protein 1, HMGB1)是全身炎癥過程中出現最晚、持續時間最長的晚期炎癥介質,與TAR4結合激活P38和NF-κB,激活的P38-MAPK和NF-κB可促進MIF的釋放,進而阻止巨噬細胞往細胞損傷部位移動,炎癥反應持續進展,進一步加重肝損傷[18](圖1)。
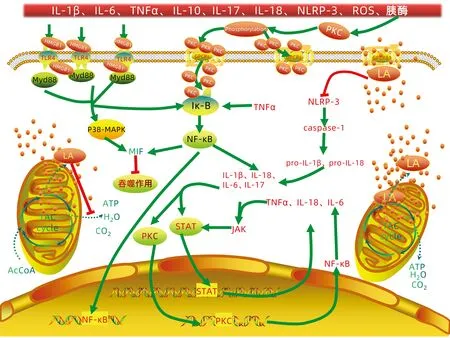
注:AcCoA,乙酰輔酶A;ATP,三磷酸腺苷;TAC cycle,三羧酸循環;LA,乳酸。
4 氧化應激
炎癥反應過程中,必定伴隨著細胞內部氧化-抗氧化水平的失衡,炎癥反應與氧化損傷相互影響。氧化應激反應是指細胞抗氧化系統與氧化系統的失衡,產生過量的ROS不斷在細胞內蓄積,超氧化物歧化酶(superoxide dismutase, SOD)、過氧化氫酶(catalase, CAT)、谷胱甘肽過氧化物酶(glutathione peroxidase, GST)等是氧自由基(oxygen free radical, OFR)的特異性清除酶,急性胰腺炎所產生大量的胰酶、細胞因子進入肝臟,抑制SOD、CAT、GST的活性,正常的氧化反應受損,損傷肝細胞呼吸鏈,使細胞處于缺氧狀態,能量代謝發生障礙,使無害的氧轉變為有害的OFR。隨著OFR在細胞內的過量積累,打破細胞的穩態系統后破壞細胞膜、線粒體,使細胞失去能量支撐,最終走向自溶。同時OFR還能夠損傷血管壁,發生漏血、滲液,導致水腫與出血。但在急性胰腺炎過程中胰腺釋放的各種炎癥介質使線粒體處于持續的高負荷狀態,損傷線粒體呼吸鏈。線粒體功能障礙ROS過量蓄積在急性胰腺炎肝損傷的發生、發展過程中起著至關重要的作用[27]。線粒體功能障礙使MAPK及AKT活性受到抑制,無法產生維持細胞生命活動的足夠ATP,加重ROS的過度蓄積[28]。過量產生的ROS與胰腺釋放的促炎細胞因子經門靜脈系統進入肝臟,激活肝細胞凋亡信號調節激酶1(apoptosis signal regulated kinase 1, ASK1)、應激狀態下活化JNK,促使P38-MAPK磷酸化,啟動肝細胞凋亡程序[28],同時誘導肝細胞產生大量如IL-1β、IL-6、TNFα等炎性細胞因子,加重肝臟炎癥損傷[29]。氧應激誘導型血紅素加氧酶-1(oxygen stress inducible heme oxygenase-1, HO-1)是一種機體保護性抗氧化酶,在應激狀態下,能通過抑制IL-10、TNFα等細胞因子而有效調節全身炎癥反應,從而減輕胰腺炎過程中肝臟的損傷[30]。核因子E2相關因子2(subcellular localization of nuclear factor E2-related factor 2, Nrf2)是細胞氧化應激反應中的關鍵因子,也是下游抗氧化基因表達的主要調節因子,在氧化應激過程中內源性Nrf2-ARE(抗氧化反應元件)途徑被激活,從而增加HO-1的活性(圖2),對肝細胞具有一定的保護作用,但是當損傷超過了內源性Nrf2-ARE的抗氧化能力時,這種保護作用將失效[31]。
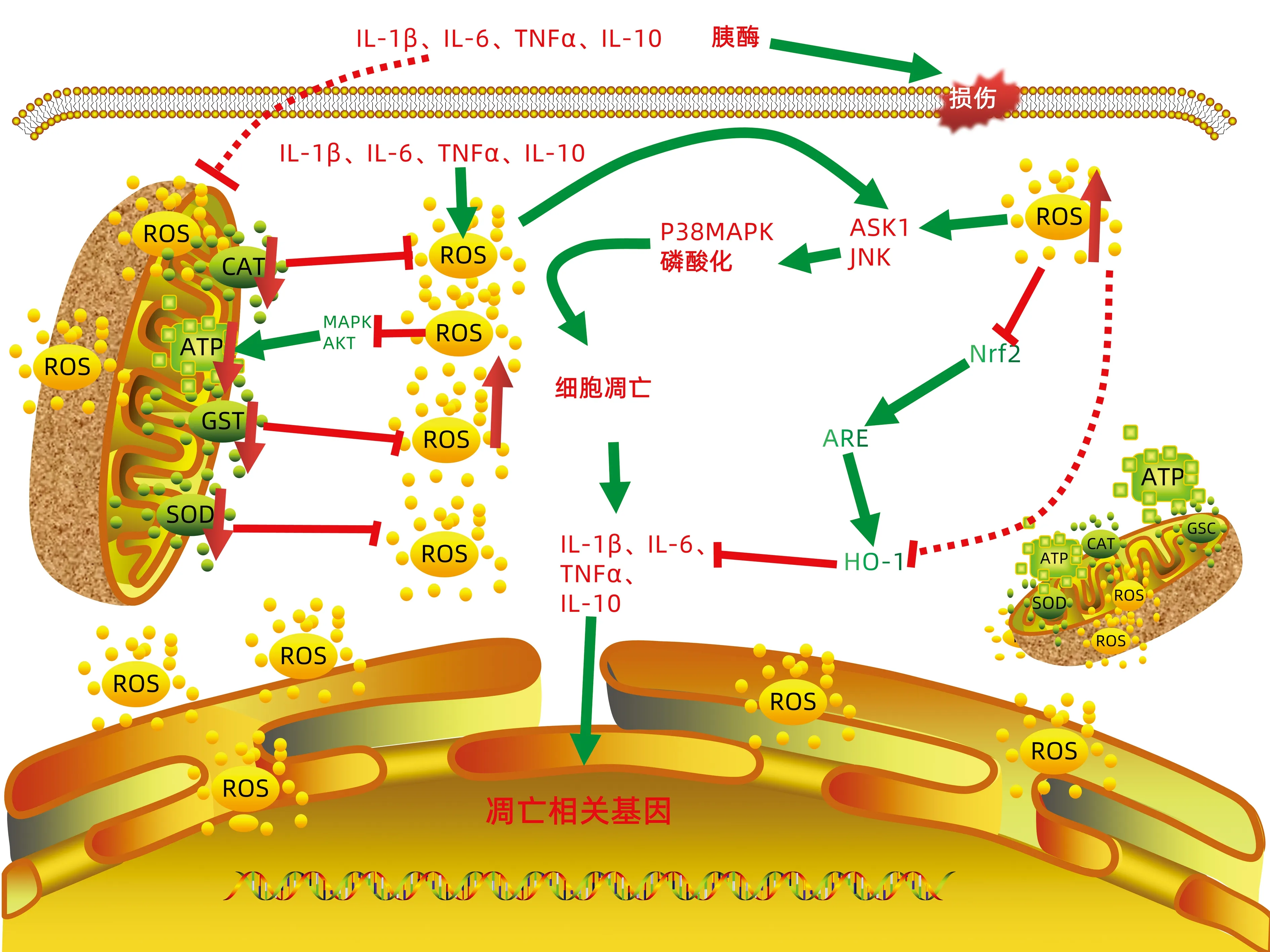
圖2 急性胰腺炎肝損傷中氧化應激分子機制
5 微循環障礙
血管是細胞因子運輸的重要通道,大量的細胞因子極易引起內皮細胞的氧化應激及炎癥反應,繼而導致破壞內皮細胞的完整性等相關問題,引起肝臟微循環障礙。血漿內皮素(endothelin, ET)和一氧化氮(nitric oxide, NO)是參與胰腺炎肝損傷微循環障礙的重要因子,生理狀態下ET、NO處于動態平衡中,維持胰腺、肝臟血流和灌注的正常狀態[32]。肝臟擁有人體最豐富的血液供應系統,血管上有豐富的ET受體,進入肝臟的胰酶、細胞因子促進ET原轉錄和ET的釋放,ET與血管上的ET受體結合,激活第二信使環磷酸鳥苷(guanosine cyclic phosphate, cGMP)途徑,細胞內Ca2+增高。細胞內過量的Ca2+會導致ATP產生減少和高電導線粒體通透性轉換孔道的持續打開,誘導細胞凋亡;Ca2+通過刺激磷酸甘油酯和α-酮戊二酸脫氫酶等催化酶可以直接促進線粒體活性氧(reactive oxygen species mitochondrion, mROS)的形成,也可以通過促進一氧化氮合酶(nitric oxide synthase, NOS)活化,形成NO,阻斷復合物Ⅳ,導致mROS過度形成,造成細胞的過氧化損傷[33]。基質金屬蛋白酶是一種膠原蛋白水解酶,參與毛細血管基底膜的降解,與ROS發揮雙重損傷作用,導致毛細血管通透性進一步增加,毛細血管滲出、水腫[34]。在急性胰腺炎時ET與NO的比值升高,使ET的縮血管作用占優勢,微血管的收縮、痙攣使內皮細胞出現缺血性損傷;內皮細胞黏附分子表達增加,使白細胞向內皮細胞黏附,白細胞與內皮之間相互作用導致靜脈回流受阻,循環血流減慢,胰腺與肝臟微循環障礙[35]。有研究[36]證實,急性胰腺炎的動物模型應用肝素后,其血液循環障礙以及炎癥反應、組織損傷各項指標均得到了改善。肝素能夠降低肝臟靜脈回流阻力,通過改善微循環障礙來減輕肝細胞的損傷。有研究[37]證實,G蛋白偶聯膽汁酸受體(G protein coupled bile acid receptor, GPBAR1)能通過參與肝細胞NF-κB、AKT、ERK等信號通路調節微循環。GPBAR1通過抑制NLRP-3炎癥小體的活化途徑降低血清淀粉酶、脂肪酶的水平,降低IL-1β、IL-6、TNFα等細胞因子和受體相互作用蛋白-3、磷酸化的混合系激酶區域樣蛋白(phospho-mixed-lineage kinase domain-like protein,p-MLKL)壞死相關蛋白的表達水平。但是在急性胰腺炎過程中,GPBAR1對機體的保護作用受到過量細胞因子及胰酶等刺激因子的抑制,放大損傷因子對肝細胞的損傷[38]。
6 腸道菌群移位
近年來腸道微生物在各種疾病中的作用受到人們的重視,急性胰腺炎過程中胰腺釋放的各種損傷因子的刺激打破腸道的穩態平衡,使腸道有害菌群沿著“腸肝循環”移位至肝臟,對肝臟造成“二次打擊”,肝損傷進一步加重。人類的胃腸道中定植著含量豐富的微生物,由超過1014種微生物和超過500萬個基因組成一個豐富的微生物群落,被稱為腸道菌群。腸道菌群通過影響新陳代謝、調節黏膜免疫系統、促進消化以及調節腸道結構等在人體生理活動中發揮著重要作用。急性胰腺炎過程中多伴有腸道菌群失衡,有害菌群過度生長、腸道黏膜屏障受損和細菌移位到外周器官導致胰腺炎不斷進展[39]。人體腸道中一些革蘭陰性細菌可以產生毒素脂多糖。益生菌是一種革蘭陽性桿狀厭氧菌,可以將人體腸道中未消化的膳食碳水化合物發酵為短鏈脂肪酸(short chain fatty acid, SCFA),尤其是丁酸[40]。丁酸、丁酸鹽等有益菌種下降使緊密連接蛋白與SCFA水平下降,人腸黏膜通透性增高、腸黏膜屏障功能障礙,導致腸道內致病菌通過損傷的腸黏膜釋放入血,經門靜脈回流系統及腸肝毛細血管網匯聚于肝臟[41],引起肝臟感染,其分子機制可能是腸道內致病菌誘導NLRP-3炎癥小體活化加重炎癥反應,通過P38-MAPK信號通路增加肝細胞凋亡。在急性胰腺炎過程中常伴隨腸道菌群多樣性的改變,IL-22作為腸道上皮內穩態關鍵調節器在急性胰腺炎因腸道菌群移位導致多臟器損傷的過程中發揮著正向調節作用[42]。IL-22能夠通過激活STAT3信號通路增加Reg-Ⅲβ、Reg-Ⅲγ、Bcl-2、Bcl-xL等保護性基因的表達,在一定程度上減輕腸黏膜屏障的損傷,減少脂多糖釋放入血,在一定程度上減輕了急性胰腺炎過程中的腸源性肝損傷[43]。
7 總結
在急性胰腺炎過程中,肝臟作為最先受到細胞因子攻擊的臟器,肝損傷可作為急性胰腺炎多臟器損傷開始的標志,急性胰腺炎肝損傷的預后在一定程度上也反映了胰腺炎的病情走向,目前急性胰腺炎肝損傷的機制尚不明確,雖然本文分為6個方面對急性胰腺炎肝損傷機制進行闡述,但這6個方面并非獨自作用于疾病進程,而是相互聯系,相互交織,共同推動疾病進展。基于目前研究現狀,臨床上應重視肝臟、腸道與胰腺的整體性,在急性胰腺炎的基礎用藥方面,將改善微循環制劑與調節腸道菌群制劑合用于急性胰腺炎的治療中,也許有利于阻斷急性胰腺炎肝損傷及其他臟器損傷的通路,逆轉疾病進程。
利益沖突聲明:所有作者均聲明不存在利益沖突。
作者貢獻聲明:徐文倩、郭敏對研究的思路或設計有關鍵貢獻,參與研究數據的獲取分析解釋過程;吳瑤麒、張近遠參與撰寫及修改文章;李合國、王曉對文章關鍵內容進行指導與修改。
