一鍋法合成2-羥基-9-芴酮類化合物
2022-10-28 00:56:04朱啟萌姜雅坤武超芊毛亞蘭范曉語王程宇
合成化學 2022年10期
朱啟萌, 姜雅坤, 武超芊, 毛亞蘭, 范曉語, 王程宇
(臨沂大學 化學化工學院,山東 臨沂 276000)
芴酮骨架大量存在于具有生理活性的天然產物或藥物分子中,呈現出各種不同的藥效價值[1-2]。1970年,Krueger等[3]發現化合物Tiorone dihydrochloride在老鼠體內具有抗病毒藥理作用,之后研究者們陸續發現該化合物還具有增強自然殺傷細胞的吞噬活性[4]、解熱[5]、抗纖維化[6]、消炎[7]以及干擾素誘生[8]等藥理活性,該藥效作用部分歸因于芴酮骨架的平面結構修飾[9]。

Chart 1

Scheme 1
此外,芴酮類化合物由于其獨特的光電性能,也常作為可調控的合成子,應用于有機半導體光電聚合材料領域[10]。例如,聚合物PF可以作為電子注射材料[11],HIQF1可以作為太陽能電池敏化染料[12]等(Chart 1)。由于芴酮類化合物顯著的藥理活性和獨特的光電性能,其構建方法引起了有機化學家們的廣泛關注。
芴酮骨架的合成策略大致可以分為兩類:分子內反應和分子間反應。分子內反應常用的反應底物包括鄰羰基聯苯類化合物[13-14]、二苯甲酮衍生物[15-17]和三炔類化合物[18]等,通過傅克酰基化、氧化環化、過渡金屬催化分子內C—H鍵活化和[4+2]環加成等反應模式均可構建芴酮分子。2006年,Langer等[13]報道了通過多步反應合成鄰甲酸酯聯苯類化合物,之后在酸性條件下發生分子內傅克酰基化反應得到芴酮衍生物;2013年,Glorius課題組[14]報道了鄰甲酰基聯苯類化合物在過氧化物存在條件下,底物中甲酰基中的C—H鍵均裂產生酰基自由基,進而分子內自由基環化得到芴酮骨架;2018年,Hoye等[18]報道了鄰共軛雙炔芳基炔酮底物在酸性條件下發生分子內[4+2]反應得到芴酮類化合物。然而,這些方法往往需要多步合成反應底物,且反應體系多涉及強酸、過氧化物和較高溫度等不利因素。
分子間反應常見的反應模式包括:(1) 2-鹵聯苯類化合物與CO[19]、甲酸苯酯[20]等,過渡金屬催化插羰環化反應。2002年,Larock課題組[19]報道了2-鹵聯苯類化合物在Pd催化下氧化加成得到C-PdX物種,進而與CO發生插羰環化反應得到芴酮衍生物。(2)鄰鹵苯甲醛與芳基硼酸[21]、鹵代芳基硼酸[22]以及苯炔前體[23]等,偶聯環化反應。2010年,Ray等[22]報道了一種在過渡金屬催化下鄰溴苯甲醛與鄰溴芳基硼酸經分子間偶聯環化反應構建芴酮類化合物。該反應首先發生Suzuki偶聯反應得到鄰(2-鹵苯基)苯甲醛類化合物,進而經過氧化加成、分子內插羰、還原消除得到相應產物。(3)含導向基團的芳基化合物與鹵代芳烴[24]、芳基硼酸[25]等,分子間C—H鍵活化構建芴酮骨架,導向基團可以為肟醚、酰胺、胺、氰基、羧酸和醛基等(Scheme 1)。2010年,Shi課題組[25]報道了芳基肟醚與芳基硼酸在過渡金屬催化下經分子間反應構建芴酮衍生物。該反應以肟醚為導向基團,在Pd催化下形成環Pd中間體,進而與芳基硼酸金屬交換,還原消除得到鄰苯基芳基肟醚中間體,再經進一步環化水解得到相應產物。2013年,Hsieh課題組[26]報道了鄰氰基聯苯類化合物與鹵苯分子間串聯環化反應合成芴酮類化合物。該反應以氰基作為導向基團,經C—H鍵活化、氧化加成和還原消除得到鄰氰基聯苯類化合物,該化合物再經進一步環化,水解得到相應產物。然而這些方法往往涉及貴金屬催化和復雜配體使用,且部分反應的收率不高。因此,發展廉價、溫和高效的芴酮骨架構建方法十分必要。

Scheme 2

Scheme 3
本文設計并合成了不同取代類型的鄰呋喃芳基炔酮底物(1),并利用其分子內呋喃環與炔鍵的D-A反應,在酸性催化條件下環氧開環并發生芳構化,一鍋法合成2-羥基-9-芴酮衍生物(2a~2e, Scheme 2),收率89%~96%,化合物2a~2e結構經1H NMR,13C NMR和HR-MS(ESI)表征。最后,將合成的產物2e作為底物按照文獻方法[27]完成了化合物Tilorone的合成(Scheme 3)。
1 實驗部分
1.1 儀器與試劑
Bruker Avance III 400 MHz型核磁共振儀;Agilent 6540 Q-TOF型高分辨質譜儀。
不同取代類型的鄰溴苯甲醛、三丁基呋喃錫、氟化鉀、不同類型的末端炔、正丁基鋰、鄰碘酰苯甲酸(IBX)、甲苯、一水合對甲苯磺酸等。所有試劑均為分析純。
1.2 合成
(1) 鄰呋喃芳基炔酮底物(1a)的合成
在氮氣氛圍下向反應管中依次加入鄰溴苯甲醛1.16 mL(10.00 mmol)、甲苯(20.00 mL)、三丁基呋喃錫3.50 mL(11.00 mmol)、 Pd(dba)2115.00 mg(0.10 mmol)和PPh3210.00 mg(0.80 mmol),封管110 ℃條件下反應3~5 h(TLC檢測)。待反應原料消失后,加入飽和氟化鉀溶液淬滅反應,有大量沉淀產生。反應液經硅藻土過濾,濾液用乙酸乙酯(3×20.00 mL)萃取,有機相用飽和食鹽水洗滌(30.00 mL),無水硫酸鈉干燥,減壓濃縮,殘余物經硅膠柱層析(洗脫劑:乙酸乙酯 ∶石油醚=1 ∶25,V∶V)純化得化合物鄰呋喃苯甲醛化合物1.49 g,收率86%。
氮氣氛圍下,在干燥的Schlenk反應管中依次加入苯乙炔1.52 mL(13.79 mmol)和無水THF(15.00 mL)。將反應管置于-78 ℃條件下,加入正丁基鋰5.20 mL(12.93 mmol)并在該條件下保持30 min。將上一步合成的鄰呋喃苯甲醛化合物1.48 g(8.62 mmol)溶于無水THF(5.00 mL)中,并注入反應體系。之后將反應溫度降至室溫并反應1~3 h(TLC檢測)。反應結束后,加水(30.00 mL)淬滅,乙酸乙酯(3×20.00 mL)萃取,有機相用飽和食鹽水洗滌(30.00 mL),無水硫酸鈉干燥,減壓濃縮得到鄰呋喃芳基炔醇粗產品,并直接用于下一步反應。
空氣氛圍下,用15.00 mL二甲亞砜分2~3次將上一步產品轉移至100.00 mL圓底燒瓶中。加入鄰碘酰苯甲酸(IBX) 2.90 g(10.34 mmol, 1.20 eq),室溫過夜反應(TLC檢測)。反應結束后加水淬滅,產生大量白色沉淀。反應液經硅藻土過濾,濾液用乙酸乙酯(3×20.00 mL)萃取,有機相用飽和食鹽水洗滌(30.00 mL),無水硫酸鈉干燥,減壓濃縮,殘余物經硅膠柱層析(洗脫劑:乙酸乙酯 ∶石油醚=1 ∶10,V∶V)純化得鄰呋喃芳基炔酮化合物1a2.00 g,收率85%,棕色固體。用類似方法合成反應底物1b~1e,收率83%~95%。
(2) 2-羥基-9-芴酮衍生物(2)的合成
空氣氛圍下向封管中依次加入反應底物1a82.00 mg(0.30 mmol)、甲苯(3.00 mL)和一水合對甲苯磺酸5.70 mg(0.03 mmol),并在封管100 ℃油浴條件下反應8~17 h(TLC檢測)。反應結束后,加入10.00 mL乙酸乙酯稀釋并轉移至100.00 mL茄形瓶,減壓濃縮,殘余物經硅膠柱層析(洗脫劑:乙酸乙酯 ∶石油醚=1 ∶ 4,V∶V)純化得2-羥基-9-芴酮化合物77.60 mg,收率95%,暗紅色固體。用類似方法合成2a~2f。
2a:暗紅色固體,收率95%, m.p.194~196 ℃;1H NMRδ: 5.32(s, 1H), 7.07(d,J=8.0 Hz, 1H), 7.17(d,J=3.2 Hz, 1H), 7.37~7.43(m, 5H), 7.47~7.54(m, 5H);13C NMRδ: 119.526, 120.34, 120.84, 124.06, 127.61, 127.92, 128.98, 129.15, 129.62, 131.21, 131.60, 134.14, 134.63, 137.45, 144.05, 154.15, 192.91; HR-MS(ESI)m/z: Calcd for C19H12O2{[M+H]+}273.0916, found 273.0920。
2b:深紅色固體,收率94%, m.p.206~207 ℃;1H NMRδ: 7.09(d,J=8.0 Hz, 1H), 7.27~7.31(m, 3H), 7.36~7.39(m, 4H), 7.66(d,J=8.0 Hz, 1H), 7.80(d,J=1.6 Hz, 1H), 10.03(s, 1H);13C NMRδ: 120.11, 120.44, 121.77, 124.92, 127.28, 127.43, 128.92, 130.03, 131.56, 132.02, 133.09, 133.97, 139.80, 145.87, 156.86, 190.99; HR-MS(ESI)m/z: Calcd for C19H11ClO2{[M+H]+}307.0520, found 307.0512。
2c:紅色固體,收率90%, m.p.210~213 ℃;1H NMRδ: 6.97~7.02(m, 1H), 7.09(d,J=8.0 Hz, 1H), 7.30~7.34(m, 2H), 7.35~7.41(m, 3H), 7.43(dd,J1=8.2 Hz,J2=5.6 Hz, 1H), 7.56(dd,J1=9.0 Hz,J2=2.0 Hz, 1H), 7.62(d,J=8.0 Hz, 1H), 10.02(s, 1H);13C NMRδ: 107.73(d,J=24.6 Hz), 114.01(d,J=23.4 Hz), 120.27, 121.66, 125.84(d,J=10.4 Hz), 127.22, 127.27, 128.78, 129.82, 130.06, 131.85, 133.12, 133.76, 147.12(J=10.6 Hz), 156.84, 166.85(d,J=250 Hz), 190.65; HR-MS(ESI)m/z: Calcd for C19H11FO2{[M+H]+}291.0816, found 291.0808。
2d:紅色固體,收率89%, m.p.264~265 ℃;1H NMRδ: 3.77(s, 3H), 3.92(s, 3H), 6.92(s, 1H), 6.95(d,J=8.0 Hz, 1H), 7.24(s, 1H), 7.28~7.40(m, 6H), 9.61(s, 1H);13C NMRδ: 55.67, 55.96, 103.43, 106.75, 119.11, 119.63, 125.66, 126.86, 126.98, 128.57, 130.01, 131.78, 133.41, 134.90, 139.02, 148.28, 154.58, 155.36, 191.32; HR-MS(ESI)m/z: Calcd for C21H16O4{[M+H]+}333.1121, found 333.1117。
2e:紅色固體,收率96%, m.p.269~271 ℃;1H NMRδ: 8.77(s, 2H), 7.40(d,J=8.0 Hz, 2H), 7.01(d,J=2.4 Hz, 2H), 6.95(dd,J1=8.0 Hz,J2=2.4 Hz, 2H);13C NMRδ: 193.96, 158.62, 137.50, 136.83, 121.76, 121.73, 111.97; HR-MS(ESI)m/z: Calcd for C13H8O3{[M+H]+}213.0552, found 213.0556。
(3)Tilorone的合成
按文獻報道的方法[27],以2,7-二羥基芴酮(2e)為反應原料,完成了化合物Tilorone的合成。在反應管中依次加入2-氯-N,N-二乙基二胺鹽酸鹽(95.50 mg, 0.56 mmol, 3.70 eq)、 2,7-二羥基芴酮(2e)(31.80 mg, 0.15 mmol, 1.00 eq)、 KOH(71.50 mg, 1.28 mmol, 8.50 eq)、甲苯(1.20 mL)和H2O(0.30 mL)。將反應體系置于110 ℃油浴條件下回流加熱24 h(TLC檢測)。反應結束后,加水(30.00 mL)淬滅反應,乙醚(3×15.00 mL)萃取,合并有機相并用無水硫酸鈉干燥。減壓濃縮,殘余物經硅膠柱層析(硅膠預先用5% NEt3二氯甲烷溶液浸泡)(洗脫劑:二氯甲烷 ∶甲醇=20 ∶1,V∶V)純化得化合物Tilorone40.00 mg,收率65%,橙色固體。1H NMRδ: 1.07(t,J=7.1 Hz, 12H), 2.64(q,J=7.1 Hz, 8H), 2.87(t,J=6.1 Hz, 4H), 4.06(t,J=6.1 Hz, 4H), 6.94(dd,J1=8.1 Hz,J2=2.5 Hz, 2H), 7.15(d,J=2.4 Hz, 2H), 7.25~7.28(m, 2H);13C NMRδ: 12.01, 48.00, 51.73, 67.23, 110.49, 120.62, 120.91, 136.06, 137.60, 159.40, 193.94; HR-MS(ESI)m/z: Calcd for C25H35N2O3{[M+H]+}411.2648, found 411.2698。
2 結果與討論
2.1 反應條件優化
以10% TsOH·H2O為催化劑,DCM為反應溶劑,在封管100 ℃條件下反應15 h。TLC檢測反應結束,直接旋干過柱,以86%的收率得到2-羥基-9-芴酮化合物2a,產物結構經核磁共振氫譜、碳譜以及X-ray單晶衍射確證(CCDC: 2144829)[18](圖1)。之后在反應催化劑為10% TsOH·H2O、反應溫度為100 ℃的條件下,考察不同極性大小溶劑如DMF、 1,4-dioxane和toluene等對反應效率的影響(表1, Entry 2~4),結果表明,toluene作為反應溶劑時的效果最好(Entry 4)。隨后進一步考察了在反應溶劑為toluene,反應溫度為100 ℃的條件下,不同類型的路易斯催化劑如FeCl3、 ZnCl2、 AlCl3和BF3·Et2O等對反應效率的影響(表1, Entry 5~8)。由表1可以看出,這些常見的路易斯酸催化劑均能較好地催化反應并能順利得到目標產物,收率70%~93%。此外,在不加催化劑的情況下反應19 h,以60%收率得到產物(Entry 9)。而當降低反應溫度(Entry 10)或減少TsOH·H2O催化劑用量(Entry 11)時,產物收率均有不同程度下降。因此,該反應的最佳反應條件為:10% TsOH·H2O為反應催化劑,toluene為溶劑,反應溫度為100 ℃。

圖1 化合物2a的單晶結構Figrue 1 Single crystal structure of compound 2a
2.2 反應底物拓展
在最佳反應條件下,通過改變鄰呋喃芳基炔酮底物1中R1、 R2取代基進行反應普適性考察。研究結果表明,母環上取代基R1無論是吸電子基(4-Cl、 4-F)還是供電子基(4,5-二甲氧基、5-OH),均能以較優收率得到2-羥基-9-芴酮產物(89%~94%)。 R2無論是芳基炔還是末端炔,同樣也能順利得到相應產物(95%~96%)。因此,該反應的普適性良好。

表1 反應條件優化
Scheme 4
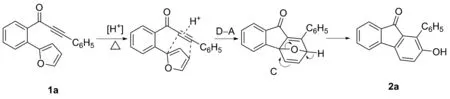
2.3 反應機理
該反應的可能反應機理如Scheme 4所示。反應底物1a在催化劑及加熱條件下,發生分子內D-A反應得到氧雜橋環化合物C,化合物C進而在酸性催化條件下發生環氧開環、芳構化過程得到2-羥基-9-芴酮化合物2a。
本文發展了一種合成2-羥基-9-芴酮類化合物的方法,以鄰呋喃芳基炔酮為反應底物,利用其分子內D-A反應、環氧開環和芳構化反應,一鍋法合成了5種新型的2-羥基-9-芴酮衍生物,收率89%~96%。該方法原料易得,催化體系簡單,低成本,反應條件相對溫和,且產物中2-位含羥基,便于進一步反應獲得衍生物。
