在線固相萃取-超高效液相色譜-串聯質譜法測定水體中氯胺酮、去甲氯胺酮和羥亞胺
2022-10-28 07:01:22朱思琪徐柏楊諸葛偉偉何穎聲
中國司法鑒定 2022年5期
朱思琪,徐柏楊,諸葛偉偉,何穎聲
(國家毒品實驗室浙江分中心(浙江省毒品技術中心)浙江省禁毒和毒情監測關鍵技術研究重點實驗室,浙江 杭州 310053)
氯胺酮于1962年首次作為麻醉劑被合成,2003年我國公安部已將其列入毒品范疇。利用合法途徑限制性使用氯胺酮是世界人民的共同期望,但目前依然有大量氯胺酮作為毒品被濫用。氯胺酮易導致精神分裂和致幻作用,不僅使吸食者生命健康受到損害,甚至會造成極其惡劣的社會危害,故應對其密切關注。去甲氯胺酮是氯胺酮在生物體內的代謝產物,在吸食氯胺酮人群的毛發、尿液及生活污水中均能檢出。羥亞胺是氯胺酮的同分異構體,也是合成氯胺酮的前體,通過簡單加熱即可重排得到氯胺酮,并且無論通過哪一種合成路徑,最終都是以羥亞胺作為氯胺酮合成的最終前體。2008年,羥亞胺作為一類易制毒化學品被嚴格管理。為了發現潛在的制毒工廠,排摸隱性吸毒人員,評價地區毒情形勢,監測環境中的氯胺酮,有必要同時對氯胺酮、去甲氯胺酮及其前體羥亞胺進行有效監測。
污水流行病學(Wastewater-Based Epidemiology,WBE)基于對未經處理的廢水中毒品及其代謝物含量的測定來統計區域毒品消費情況,成為毒品使用情況的有效監測手段。針對城市生活污水中毒品及其代謝物的研究較多,但是對于相關易制毒化學品的監測報道相對較少。羥亞胺的分析通常是基于氣相色譜-質譜聯用法(Gas Chromatography-Mass Spectrometry,GC-MS)、氣相色譜法(Gas Chromatography,GC)或液相色譜-紫外光譜法(Liquid Chromatography-Ultra Violet,LC-UV)等分析方法。由于氣相色譜儀進樣口的溫度及襯管的惰性程度均會影響其穩定性,難以避免一部分羥亞胺轉變為氯胺酮,因此無法實現氯胺酮和羥亞胺的準確測定。LC-UV可滿足氯胺酮和羥亞胺的同時準確測定,但其檢出限僅為5 mg/L,難以滿足實際水體中檢測的需求。近年來,由于液相色譜-串聯質譜聯用法(Liquid Chromatography-Tandem Mass Spectrometry,LC-MS/MS)的測定靈敏度高,已廣泛應用于污水中毒品及其代謝物的研究。周爽等首次建立了羥亞胺的LC-MS/MS分析方法,具有較好的穩定性和準確度,可用于氯胺酮和羥亞胺的同時測定,檢出限可達1 μg/L。ZHANG等建立了離線固相萃取-液相色譜-串聯質譜聯用法(Solid Phase Extraction-Liquid Chromatography-Tandem Mass Spectrometry,SPE-LCMS/MS),實現了環境水樣中甲基苯丙胺、苯丙胺、氯胺酮、麻黃堿和羥亞胺的同時測定,并對其發生、分布以及可能存在的環境危害進行了詳細研究。
由于水樣中氯胺酮及相關物質含量較低,且基質較為復雜,一般需要輔以樣品前處理技術進行凈化和富集,采用離線固相萃取技術過程較為繁瑣,所需分析時間較長。為更好地對氯胺酮等相關物質進行快速高靈敏的分析,本文擬建立在線固相萃取-超高效液相色譜-串聯質譜法(Solid Phase Extraction-Ultra High Performance Liquid Chromatography-Tandem Mass Spectrometry,SPE-UPLC-MS/MS)快速分析污水中氯胺酮及相關代謝物和制毒前體的新方法,并將其應用于浙江省大型污水處理廠和產業園區內污水樣品的測定。
1 材料與方法
1.1 儀器與試劑
Waters ACQUITY Xevo TQ-XS型在線固相萃取液質聯用儀(美國Waters公司)。
實驗樣品A~A來源于浙江省六個地級市的大型污水處理廠污水;實驗樣品B~B來源于浙江省產業園區污水。使用全自動采樣器進行24 h混合樣品采集,每個點位選擇一個工作日和一個休息日,取樣時間為2021年6—9月。
質量濃度為1 mg/mL的氯胺酮、去甲氯胺酮、羥亞胺標準品溶液,質量濃度為100 μg/mL的內標氯胺酮-D、去甲氯胺酮標準品溶液-D,(美國Cerilliant公司);甲醇、乙腈、甲酸均為色譜純及以上;去離子水由Milli-Q超純水系統制備。
1.2 方法
1.2.1 儀器工作條件
在線固相萃取條件Waters Oasis HLB固相萃取柱(2.1 mm×30 mm,20 μm);進樣量為500 μL;流動相A為水,流動相B為乙腈,四元泵用于在線固相萃取,具體條件見表1。

表1 四元泵工作條件
色譜條件Waters ACQUITY UPLC HSS T色譜柱(2.1 mm×100 mm,1.8 μm);柱溫40℃;流動相A為0.1 %(體積分數,下同)甲酸水溶液,流動相B為乙腈,二元泵梯度洗脫程序見表2。
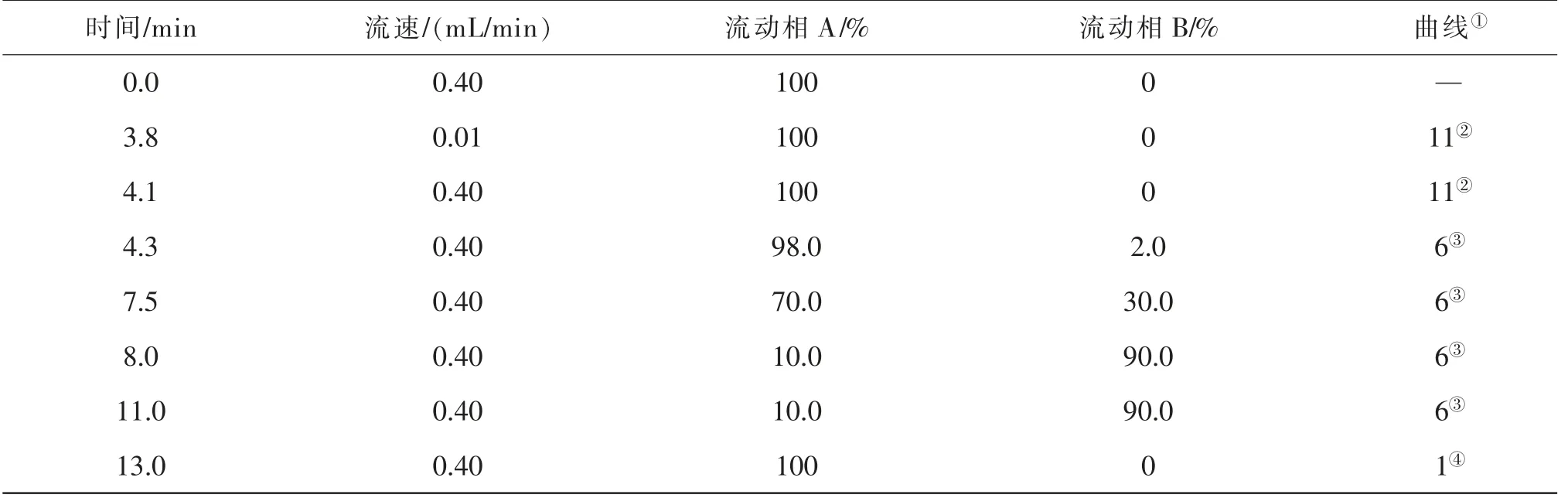
表2 二元泵梯度洗脫程序
質譜條件 電噴霧離子源(Electron Spray Ionization,ESI),正離子模式(ESI),多反應監測(Multiple Reaction Monitoring,MRM)模式;離子源溫度600℃;噴霧電壓0.5 kV;各目標物的MRM優化參數見表3。
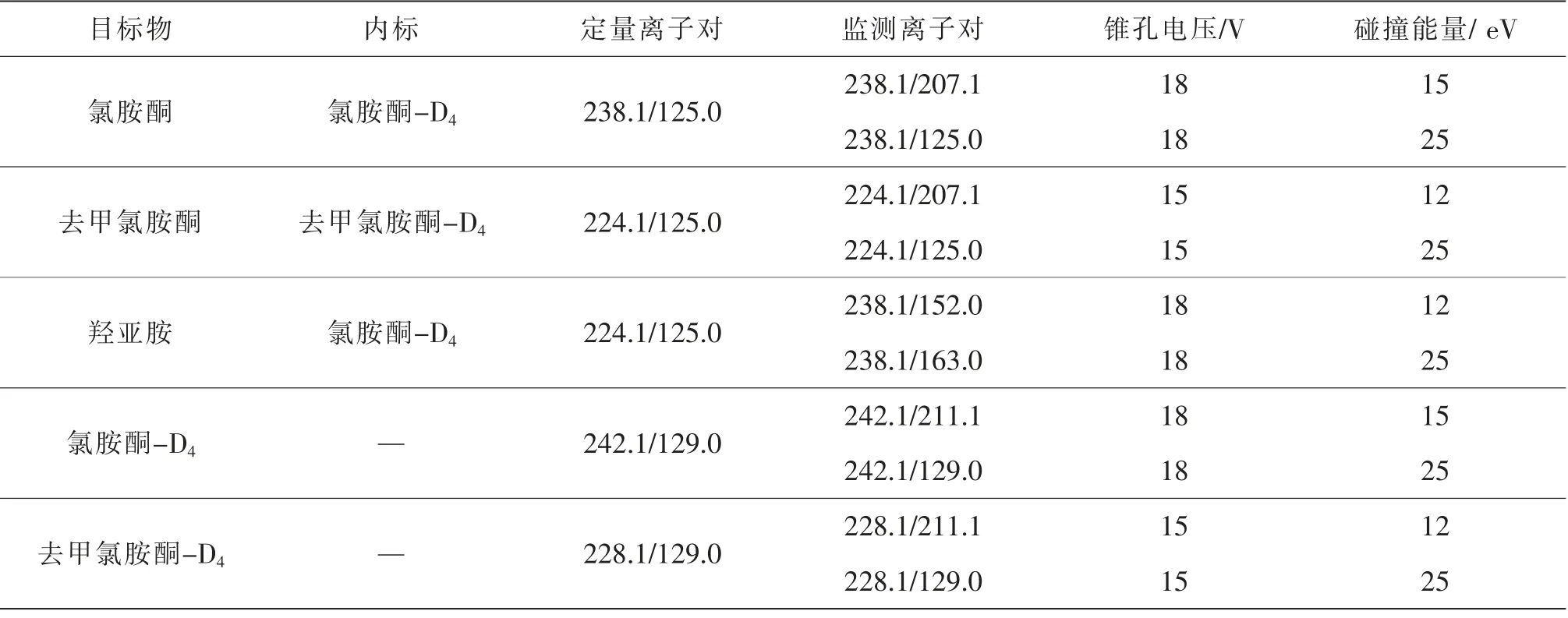
表3 各目標物的MRM優化參數
1.2.2 樣品處理過程
取樣品經0.45 μm濾膜過濾,取濾液50 mL轉移至離心管中,加入質量濃度為25 ng/mL的氘代內標混合溶液100 μL,渦旋混勻,用0.22 μm濾膜過濾后待測。每份樣品平行操作兩次,空白溶液取超純水50 mL與樣品同法操作。
1.3 方法學驗證
1.3.1 選擇性
取超純水,按已建立的方法分析考察空白水樣對目標物及內標是否存在干擾。
1.3.2 標準曲線、檢出限與定量限
按照實驗方法對各質量濃度的標準溶液進行測定,考察方法線性。以目標物和相應內標物峰面積的比值作為橫坐標(x),以目標物的質量濃度為縱坐標(y)繪制標準曲線,得到線性回歸方程及相關系數。根據國際純粹與應用化學聯合會(International Union of Pure and Applied Chemistry,IUPAC)規定,以3倍信噪比、10倍信噪比計算檢出限與定量限。
1.3.3 精密度與準確度
取自來水,分別添加不同量的混合標準儲備溶液,得到目標物質量濃度分別為2、50、150 ng/L的溶液,每個質量濃度平行測定6次,計算回收率,并以測定值的相對標準偏差作為日內精密度,連續測定3 d,計算日間精密度。
1.3.4 基質效應
分別對自來水、超純水加標至質量濃度為2、50、150 ng/L,采用公式(1)計算基質效應。
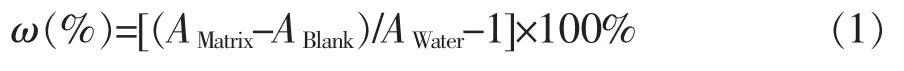
其中:A為目標物在基質中定量離子對的峰面積;A為基質空白中定量離子對的峰面積;A為目標物在超純水中定量離子對的峰面積。
1.3.5 穩定性
取50 ng/L加標樣品,分別放置0、24、48 h后進樣測定,以考察樣品的穩定性。
2 結果與討論
2.1 色譜行為
分別取質量濃度均為50 ng/L的氯胺酮、去甲氯胺酮和羥亞胺單一標準儲備溶液、質量濃度均為50 ng/L的氯胺酮-D、去甲氯胺酮-D內標溶液和空白水樣進樣。結果表明,3種目標物的分離情況良好,保留時間分別為7.88、7.72、8.23 min,基質對其他物質的定性、定量均無干擾。目標物的提取離子色譜圖如圖1所示。
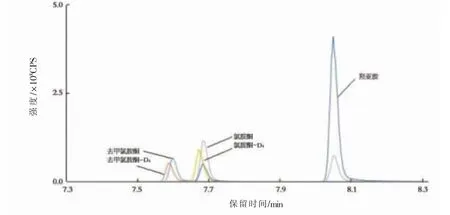
圖1 目標物的提取離子色譜圖
2.2 標準曲線、檢出限與定量限
3種目標物的線性參數、檢出限與定量限結果如表4所示。本方法對3種目標物的檢測范圍均為0.2~200 ng/L,檢出限為0.02~0.05 ng/L,定量限為0.05~0.1 ng/L,相關系數均大于0.999,可滿足實際檢測的需求。
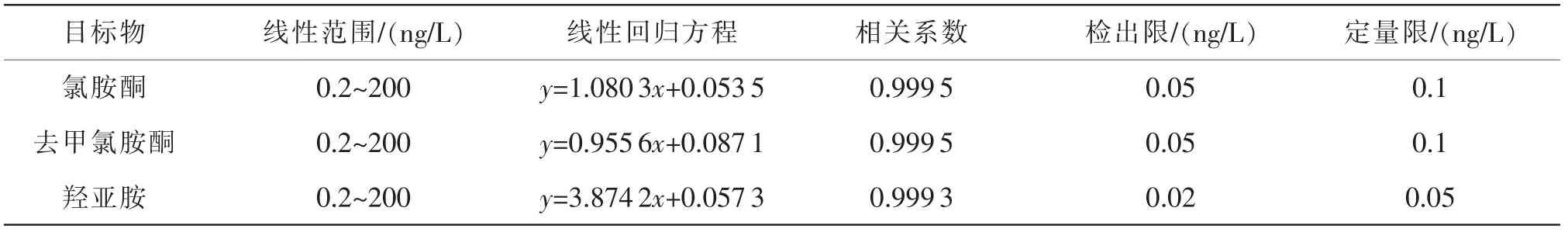
表4 線性參數、檢出限與定量限
2.3 精密度與準確度實驗
本方法對氯胺酮、去甲氯胺酮和羥亞胺的平均加標回收率分別為98.0%~101%、99.3%~100%、91.3%~102%,3種目標物在不同加標質量濃度下的日內精密度和日間精密度為0.6%~3.5%,具體結果見表5。
2.4 基質效應
結果如表5所示,3種目標物的基質效應為99.8%~114%,通過內標校正基本可消除基質效應的影響。
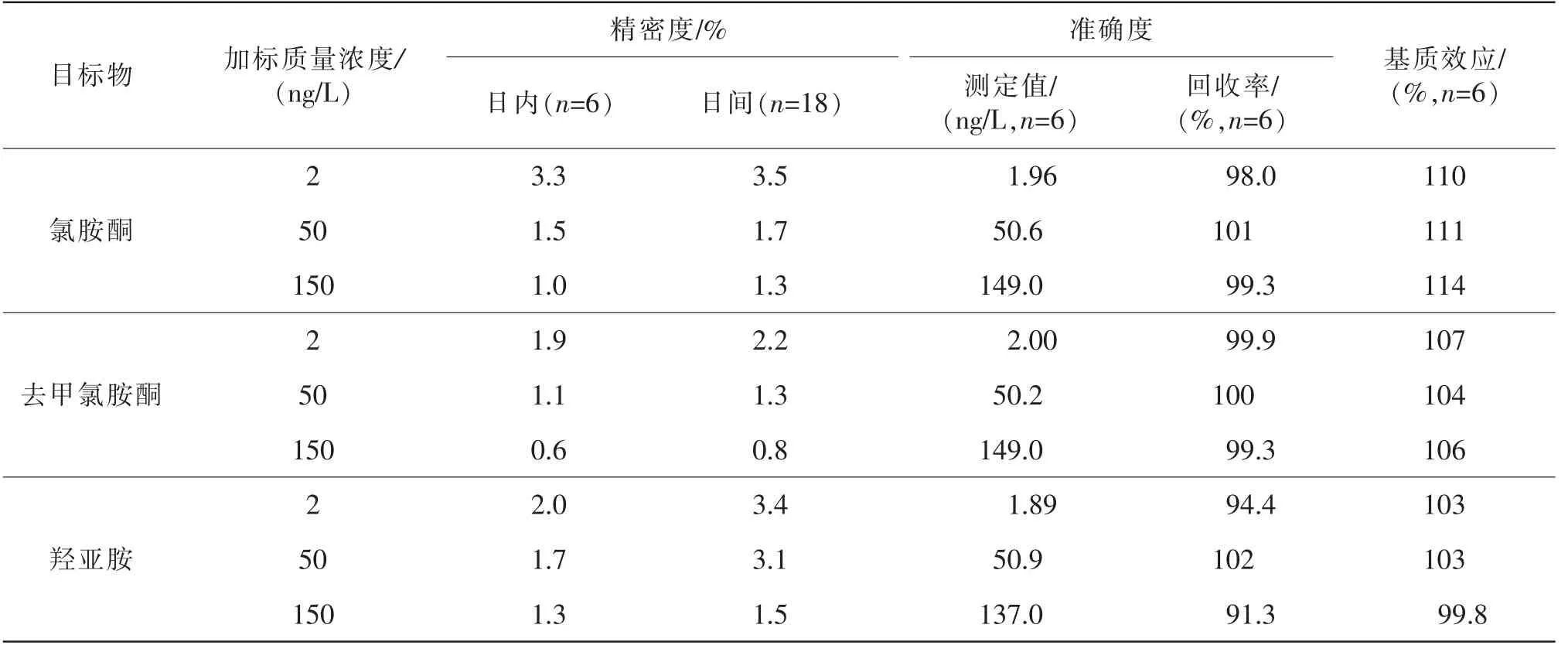
表5 精密度與回收率實驗結果
2.5 穩定性
實驗對質量濃度為50 ng/L加標樣品的穩定性進行考察。結果發現,氯胺酮和羥亞胺質量濃度的變化僅有0.7%、0.8%,去甲氯胺酮的質量濃度下降了9.0%,這表明樣品在48 h內基本穩定。
2.6 實際樣品分析
按照實驗方法對所有大型污水處理廠和產業園區污水樣品進行測定。結果表明:僅有樣品A、A、A和B中檢出氯胺酮原體,質量濃度分別為0.45、0.36、1.57、0.43 ng/L;所有樣品均未檢出去甲氯胺酮和制毒前體羥亞胺。
2.7 方法比較
目前尚未有文獻報道同時分析水體中氯胺酮、去甲氯胺酮和羥亞胺的方法。將本文方法與已報道的文獻相比,結果見表6。由此可知,本方法具有較低的檢出限,無需離線活化固相萃取柱、真空干燥萃取柱、氮吹等步驟,顯著減少了前處理步驟及分析時間。
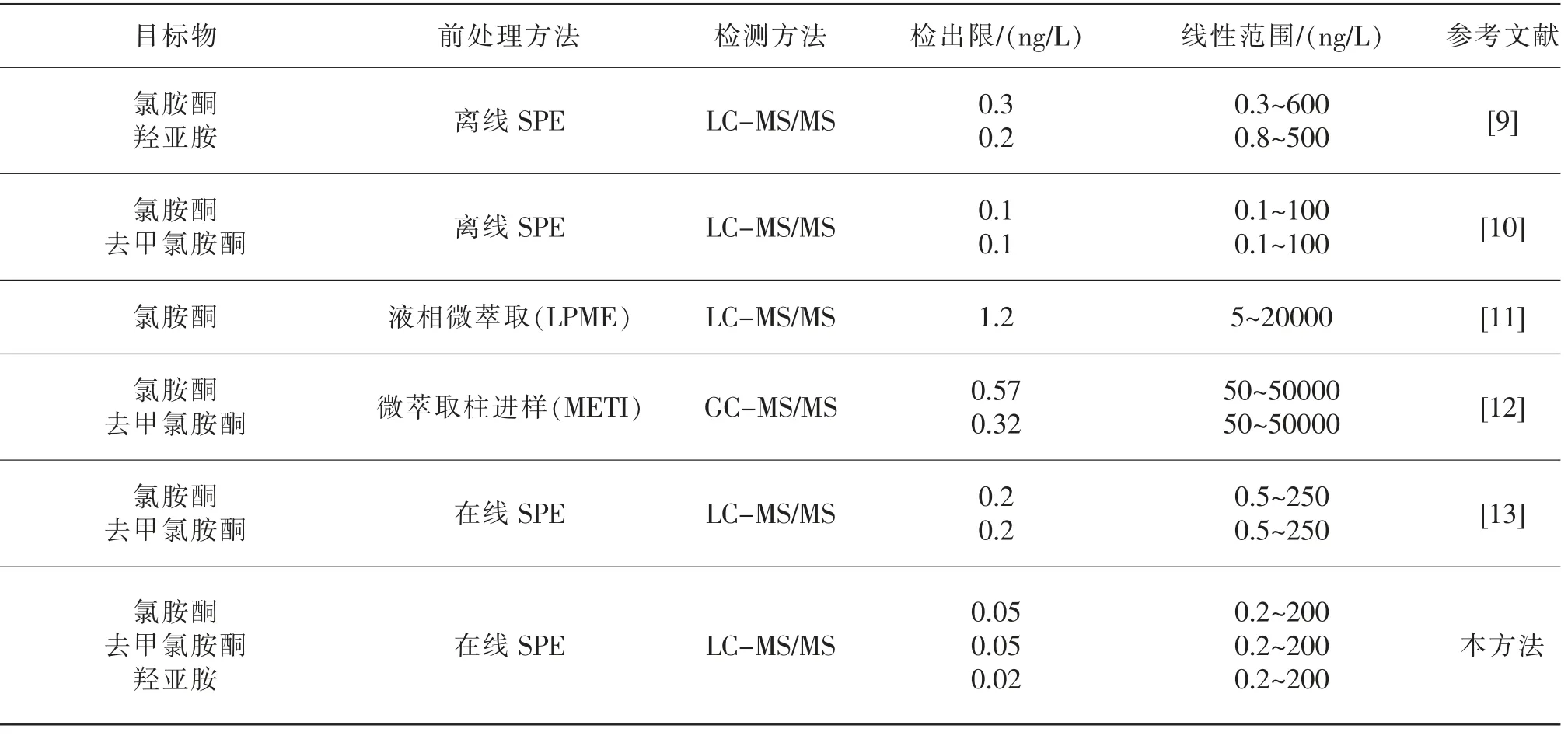
表6 不同分析方法的性能比較
3 結論
本文基于在線SPE-UPLC-MS/MS技術建立了同時快速分析水體中氯胺酮、去甲氯胺酮和羥亞胺的新方法。該方法具有較低的檢出限和較寬的線性范圍,且前處理過程較為簡單,僅需要13min即可同時實現水體中痕量氯胺酮、去甲氯胺酮和羥亞胺的測定,可成功應用于實際污水樣品的分析,為分析水體中氯胺酮及其代謝物和制毒前體提供新的監測方法。
