寡聚核苷酸介導的基因突變技術創制抗除草劑煙草新種質
2022-10-14 03:50:52謝宇峰秦利軍
廣西植物 2022年9期
關鍵詞:煙草
謝宇峰, 秦利軍
( 貴州大學 農業生物工程研究院/山地植物資源保護與種質創新省部共建教育部重點實驗室/生命科學學院, 貴陽 550025 )
煙草()為茄科(Solanaceae)煙草屬()一年生或有限多年生草本植物,是一種經濟價值較高的作物,除主要用于吸食外,煙草還具有蛋白食用及藥用價值。煙草在我國南方和北方均有種植,面積達146.67萬 hm,年均產量約30億 kg,在我國占有極大的經濟市場(張杰等,2018),是國家和地方財稅的重要經濟來源。然而,煙田雜草危害煙草生產,嚴重影響煙草的產量和質量。煙田雜草不僅與煙草爭奪水分、養分、光照和空間,而且作為煙草病蟲害傳播的中間宿主,極大影響了煙草的產量和質量(蔡海林等,2020)。在煙田中,氯磺隆、草甘膦除草劑可以通過靶向結合植物體中的乙酰乳酸合成酶(acetolactate synthase,ALS)用以抑制纈氨酸(valine,Val)、亮氨酸(leucine,Leu)、異亮氨酸(isoleucine,Ile)的生物合成,進而導致雜草死亡(Scoloss & Ciskanik, 1988)。雖然這類除草劑在殺滅雜草時具有活性高且藥量低、對哺乳動物低毒、生態環境友好的特點,但會極大影響農作物的生長和發育(Grandpmo & Peter, 1998;牛聰偉,2005)。ALS中特定氨基酸突變會使細胞系或植株產生對除草劑的抗性(Kleschick & Costales, 1990; Scoloss, 1990),如擬南芥()中ALS氨基酸保守區域第197位氨基酸由脯氨酸(proline,Pro)突變為絲氨酸(serine,Ser),會使細胞系具有對氯磺隆(chlorsulfuron,Chl)和芐嘧磺隆(bensulfuron methyl)等磺酰脲類除草劑產生抗性;甘藍()中ALS保守氨基酸序列(ahas3r)第557位Pro突變為谷氨酰胺(glutamine,Gln),會使細胞系具有對三唑嘧啶(triazole pyrimidine)類除草劑抗性(Saari & Maxwell, 1997)。此外,水稻()中ALS特定氨基酸的突變會使植株產生對除草劑的抗性。將ALS氨基酸第95位甘氨酸(glycine,Gly)突變為丙氨酸(alanine,Ala)后,獲得對雙草醚(bispyribac-sodium)產生抗性的水稻植株(Okuzaki et al., 2007)。煙草中,通過電穿孔及微粒轟擊方式將嵌合體DNA/RNA寡聚核苷酸導入原生質體,使其基因所編碼的氨基酸特定位點發生突變(脯氨酸-196-谷氨酸,色氨酸-573-亮氨酸),獲得抗氯磺隆的抗性植株(Beetham et al., 1999)。因此,利用分子生物學手段實現代謝關鍵酶編碼基因的定點突變,是創制抗除草劑作物新種質的重要手段之一。
寡聚核苷酸介導的基因突變(OMM)技術是一種新的基因誘變技術,已被成功運用于動物及酵母的基因定點突變(Yoon et al., 1996; Alexeev et al., 2000; Bartlett et al., 2000)。在植物中,該技術已成功實現對玉米()基因發生定點突變,引起AHAS蛋白中621氨基酸由Ser突變為天冬氨酸(aspartic acid,Asp),獲得對咪唑啉酮(imidazolone)類除草劑具有抗性的突變玉米植株(Zhu et al., 1999)。Okuzaki等(2007)利用OMM技術實現水稻ALS中第171位氨基酸由Pro變為Gly,第548位氨基酸由色氨酸(tryptophan,Tyr)變為Ile以及第627位氨基酸由Ser變為Ile,獲得除草劑抗性水稻。與應用廣泛的成簇規律間隔短回文重復序列及相關蛋白Cas(CRISPR/Cas)技術,以及轉錄激活樣效應因子核酸酶(TALEN)技術(Ishino et al.,1987; Michno et al., 2020)相比,OMM技術所誘導的突變幾乎都是有目的定點突變,不會向生物體內引入新的基因。Breyer等(2009)認為通過OMM技術誘導產生的生物不應當歸類于轉基因生物。在對轉基因生物安全性爭論不休的今天,OMM技術為生物品質的改良提供了一條新的途徑。本研究以煙草品種‘K326’為材料,利用同源克隆法獲得基因,根據基因保守區結構分析構建RNA/DNA嵌合體分子,并利用該嵌合體實現對基因的定點突變,創制對磺酰脲類除草劑具有抗性的煙草新種質。
1 材料與方法
1.1 材料和試劑
煙草()品種‘K326’由貴州大學農業生物工程研究院山地植物資源保護與種質創新省部共建教育部重點實驗室保存并提供。DL 2 000 DNA marker、LA Taq with GC Buffer、DNA快速純化回收試劑盒購自TaKaRa寶生物工程(大連)有限公司;MS培養基購自Phyto Technology Laboratoies公司;新型植物DNA提取試劑盒購自TIANGEN BIOTECH公司;膠回收試劑盒、pGEM-T Easy Vector System購自Promega公司;氯磺隆購自Sigma-Aldrich公司;RNA/DNA嵌合體由TaKaRa公司合成;基因克隆引物由上海英駿生物技術有限公司完成。
1.2 方法
1.2.1 煙草品種‘K326’基因組提取及基因克隆 以煙草品種‘K326’的葉片為材料,使用TIANGEN BIOTECH公司新型植物DNA提取試劑盒提取煙草基因組DNA。根據NCBI報道的普通煙草基因(基因登錄號:X07644.1和X07645.1)序列設計同源克隆引物及含588位點和1719位點突變區擴增引物,引物序列如表1所示。
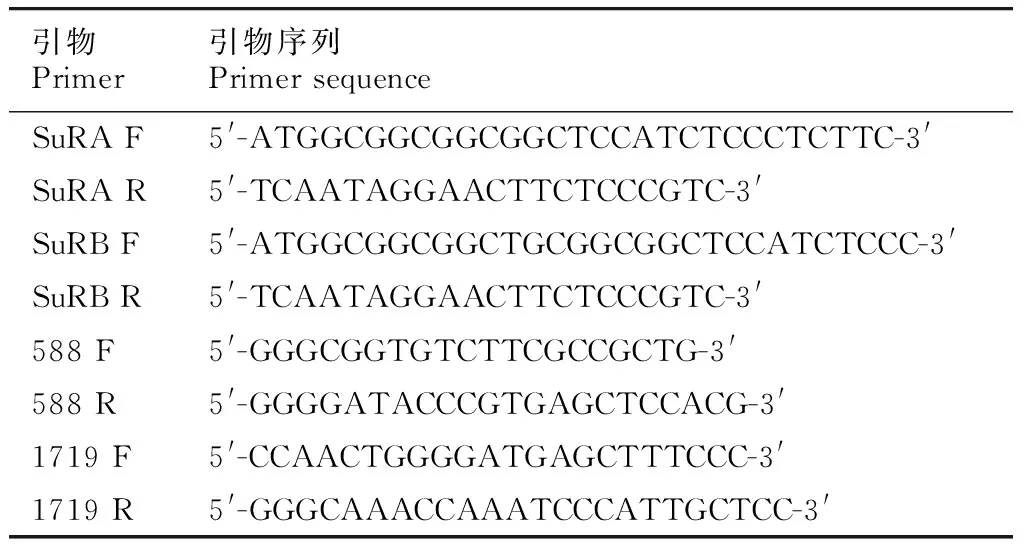
表 1 煙草ALS基因的克隆及保守區擴增引物Table 1 Cloning and conserved region amplification primer sequences of tobacco ALS gene
參照TaKaRa公司LA Taq with GC Buffer試劑盒操作方法,對基因進行擴增。擴增體系(50 μL):0.5 μL TaKaRaTaq(5 U·μL),25.0 μL 2 × GC BufferⅡ,8.0 μL dNTP Mixture(2.5 mmol·L),1.0 μL R-F(20 μmol·L),1.0 μL R-R(20 μmol·L),1.0 μL基因組DNA(100 ng)和13.5 μL ddHO。擴增條件:94 ℃預變性3 min,94 ℃變性30 s,55 ℃退火30 s,72 ℃延伸160 s,35個循環后72 ℃ 延伸 10 min。
1.2.2 PCR產物連接及測序 參照Promega公司膠回收試劑盒操作方法回收PCR產物,參照Promega公司pGEM-T Easy Vector System試劑盒說明將回收的PCR產物連接于pGEM-T Easy Vector。連接有PCR產物的pGEM-T Easy Vector導入大腸桿菌()DH5α感受態菌株。將工程菌送至北京諾賽公司測序。利用DNAMAN 9.0軟件中“序列拼接功能”對測序結果進行拼接,將拼接結果于NCBI數據庫中比對,確認保守區。
1.2.3 RNA/DNA寡聚核苷酸嵌合體構建 基于煙草品種‘K326’的基因保守區的結構,參照Kochevenko和Willmitzer(2003)的方法設計RNA/DNA嵌合體序列,交由TaKaRa公司合成。嵌合鏈設計基本要求與靶基因的序列一樣(除了突變位點外),通過寡聚核苷酸鏈代換,引入靶位點堿基序列變化。
1.2.4 基因槍介導的RNA/DNA導入煙草的愈傷組織 參照譚穎等(2013)的方法略有改動,對煙草品種‘K326’的愈傷組織進行誘導。煙草葉片經75%的酒精滅菌30 s,0.1%HgCl消毒8 min,無菌水清洗3~5次后切塊(0.5 cm×0.5 cm)接種于愈傷誘導培養皿上,經過2周誘導培養后,轉接于新鮮繼代培養基上繼續培養至出現分化芽點,分化芽接種生根培養基上生根培養。愈傷誘導培養基:MS+1.5 mg·L6-BA+0.1 mg·LNAA;繼代培養基:MS+1.0 mg·L6-BA+0.2 mg·LNAA。將煙草愈傷組織繼代培養基分組,分別加入不同濃度的氯磺隆,對愈傷組織進行氯磺隆敏感性實驗。氯磺隆濃度梯度分別設置為90、100、110、120、130、140、150、160 nmol·L。每個皿接種相同數量、生長狀態近似的愈傷組織進行繼代培養,培養條件為光照強度為2 000 lx,每日光照16 h,培養溫度為(26 ± 1)℃。培養1 周后觀察愈傷組織生長情況。抗性芽生根培養基:1/2MS + 0.1 mg·LNAA。愈傷誘導培養基和繼代培養基中補加30 g·L蔗糖和8 g·L瓊脂,生根培養基中補加30 g·L蔗糖和6 g·L瓊脂,各培養基pH均為5.8。
基因槍微彈的制備參照Bio-Rad PDS-1000He使用說明書,根據程義琳等(2013)的方法,略有改動。設計基因槍轟擊實驗流程:稱取8 mg金粉置于1.5 mL EP管中;加入1 mL無水乙醇,漩渦震蕩3~5 min,10 000 r·min離心1 min,棄上清液(重復3次);加入1 mL無菌水,震蕩1 min;加入133 μL無菌水,儲存在-20 ℃待用;加入13.3 μL嵌合體震蕩2~3 s;加入133 μL 2.5 mol·LCaCl震蕩2~3 s;加入53.3 μL 0.1 mol·Lspermidine,震蕩2~3 s;在冰上靜置10 min,10 000 r·min離心10~20 s,棄上清液;加入500 μL無水乙醇,沖洗2次,離心10 s,棄上清液;加入160 μL無水乙醇,均勻后,4 ℃備用(每次使用10 μL)。
基因槍介導的RNA/DNA導入煙草愈傷組織,參照Kochevenko和Willmitzer(2003)、程義琳等(2013)的方法,略有改動。具體操作:打開超凈臺,用70%乙醇擦拭超凈臺內部與基因槍的表面及基因槍內部;打開紫外燈,滅菌30 min,滅菌后吹風20 min;易裂片、微彈載體及阻攔網置于70%乙醇10 min,放在無菌濾紙上自然晾干;打開氦氣瓶,將壓力調制1 100 psi;將易裂片、微彈載體及阻攔網安裝并固定在基因槍真空室,將帶有煙草愈傷組織的培養皿放在托盤上,調節轟擊距離為9 cm,關閉基因槍門,打開電源及真空泵;按下真空鍵“VAC”,當真空表讀數為25 inHg時,按維持鍵“Hold”;按下“Fire”鍵,轟擊結束,按下“通氣鍵”至真空表讀數為零。打開基因槍門,取出培養皿,蓋好蓋子用封口膜封好,轉入組培室,暗恢復2 d后,氯磺隆篩選培養,溫度為(26 ± 1)℃,光照強度為2 000 lx,光照時間為 16 h。共做12批次,每批次接種25皿,每皿接種30~40個愈傷組織(d=0.5 cm),共計9 500~10 000個愈傷組織塊。
1.2.5 抗性分化植株ALS酶活的檢測 參照陳以峰等(1998)的提取方法,取移栽后3周的野生型煙草品種‘K326’葉片1 g,加入提取介質(磷酸緩沖液A)1 mL,勻漿,2 500 r·min離心20 min,取上清液即為粗酶液;取0.5 mL反應介質(磷酸緩沖液B),加入不同濃度的氯磺隆,這里設氯磺隆濃度梯度為0~500 nmol·L;加入30 μL粗酶液,總體積0.9 mL,蒸餾水補齊,37 ℃溫育1 h,加入50 μL 3 mol·L的HSO中止反應;依次加入0.5 mL 0.83%甲萘酚和0.5 mL肌酸,60 ℃溫育15 min,將溶液于2 000 r·min離心10 min,取上清液于分光光度計530 nm比色。ALS活性直接用A530 nm值來表示。以野生型煙草品種‘K326’為對照,測定抗性植株的ALS酶活。
1.2.6 抗性分化植株突變位點驗證 根據煙草品種‘K326’的基因突變位點,選擇跨該區段的引物進行擴增,擴增后進行測序,分析擬突變位點的變化情況,PCR產物擴增、連接及測序參照1.2.2。
2 結果與分析
2.1 煙草品種‘K326’的ALS基因克隆
以煙草品種‘K326’基因為模板,利用引物對SuRA F/SuRA R和SuRB F/SuRB R進行PCR擴增,均擴增得到一條長度約為2 000 bp的條帶,且擴增條帶清晰明亮。將該PCR產物分別膠回收后以pGEM-T Easy Vector進行連接,連接后的質粒載體導入大腸桿菌DH5α感受態菌株篩選陽性菌落,陽性菌落送至北京諾賽公司進行測序。測序拼接結果表明,以SuRA F/SuRA R引物擴增到序列長度為2 004 bp的基因,命名為‘ K326’,而以SuRB F/SuRB R擴增到序列長度為2 010 bp的基因,命名為‘ K326’(圖2)。
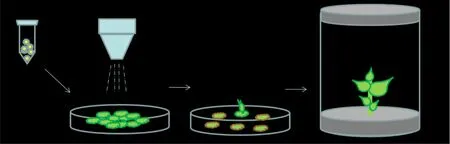
圖 1 基因槍轟擊實驗模擬流程圖Fig. 1 Flow chart of simulation by gene gun bombardment experiment

A. ‘K326’的ALS基因PCR擴增(M代表DNA 2 000 bp標記; A1-A3均代表SuRA F/SuRA R擴增產物; B1-B3均代表SuRB F/SuRB R擴增產物)。B-C. ‘K326’ALS SuRA和‘K326’ALS SuRB測序結果。A. PCR amplification of ‘K326’ALS gene ( M represents 2 000 bp Marker; A1-A3 represent SuRA F/SuRA R amplification; B1-B3 represent SuRB F/SuRB R amplification). B-C. The product sequencing of ‘K326’ ALS SuRA and ‘K326’ ALS SuRB.圖 2 煙草品種‘K326’的ALS基因擴增及PCR產物測序Fig. 2 Amplification of Nicotiana tabacum ‘K326’ALS gene and sequencing of PCR products
2.2 RNA/DNA寡聚核苷酸嵌合體設計
參照Kochevenko和Willmitzer(2003)的方法,結合‘K326’和‘K326’基因保守序列的比對,設計以‘K326’基因編碼區第588位核苷酸由胞嘧啶(cytosine,C)突變為腺嘌呤(adenine,A)的顛換,第1719位核苷酸由鳥嘌呤(guanine,G)突變為胸腺嘧啶(thymine,T)的顛換。針對編碼區核苷酸第588位點、 1719位點設計突變堿基的嵌合體分別命名為Chl-588和Chl-1719(圖3)。

大寫字母為DNA殘基; 小寫字母為2′-O-RNA殘基; 框內字母代表突變位點。Capital letters represent DNA residues; Lowercase letters represent 2′-O-RNA residues; Letters in box represent the mutation sites.圖 3 嵌合體RNA/DNA寡聚核苷酸Fig. 3 Chimeric RNA/DNA oligonucleotide
2.3 煙草品種‘K326’愈傷組織誘導及敏感性分析
通過優化的愈傷誘導培養基獲得了嫩綠色至淡黃色、結構致密的愈傷組織,可用于后續氯磺隆敏感試驗和抗性芽誘導實驗。煙草品種‘K326’愈傷組織形成后,接種于含有不同濃度氯磺隆的篩選培養基中生長1周,愈傷組織的顏色從低濃度到高濃度分組顏色由綠色變黃直至黑色(圖4)。

A. 煙草愈傷誘導及分化; B. 愈傷組織對不同濃度氯磺隆敏感性響應 (A-H分別為90、100、110、120、130、140、150、160 nmol·L-1氯磺隆)。A. Callus induction and differentiation in tobacco; B. Sensitively response of callus to different concentrations of chlorsulfonon ( A-H represent the concentration of chlorsulfonon 90, 100, 110, 120, 130, 140, 150, 160 nmol·L-1, respectively).圖 4 煙草品種‘K326’愈傷組織誘導及氯磺隆敏感性分析Fig. 4 Callus induction of tobacco‘K326’ and sensitivity analysis of chlorsulfuron
在氯磺隆濃度為90、100 nmol·L時,愈傷組織為綠色,生長狀態良好;在氯磺隆濃度為110、120、130、140 nmol·L時,愈傷組織顏色為黃色且顏色依次變暗;當氯磺隆濃度達150 nmol·L時,愈傷組織顏色呈棕色,且不繼續生長;在氯磺隆濃度達160 nmol·L時,愈傷組織呈黃黑色,逐漸發生褐化現象最后死亡。因此,本研究選擇氯磺隆濃度150 nmol·L作為篩選的臨界濃度。
2.4 基因槍轟擊處理及抗性煙苗獲得
參照程義琳等(2013)的方法,結合耿立召等(2005)和宮本賀等(2010)的方法,用包裹金顆粒的RNA/DNA寡聚核苷酸嵌合體Chl-588和Chl-1719以9 cm的轟擊距離、25 inHg的轟擊壓強對煙草品種‘K326’愈傷組織進行基因槍轟擊處理。轟擊后的愈傷組織經過恢復培養,氯磺隆篩選分化培養及抗性芽生根培養獲得具有氯磺隆抗性的煙草植株22株(圖5)。

A. 基因槍轟擊后的愈傷組織; B. 在篩選培養基上培養的愈傷組織; C. 篩選出抗性芽; D. 抗性芽生根培養; E. 獲得的抗性苗。A. Callus after gene gun bombardment; B. Callus cultured on screening medium; C. Screening resistant buds; D. Root cultivation with resistant buds; E. Resistant plants obtained.圖 5 煙草品種‘K326’基因槍遺傳轉化體系Fig. 5 Process of gene gun gemetic transformed tobacco ‘K326’
2.5 抗性煙苗的ALS酶活性測定
通過對野生型煙草品種‘K326’的ALS酶活性進行不同濃度氯磺隆敏感性檢測發現,在氯磺隆濃度為300~500 μmol·L時, ALS酶活性保持最低,在小于300 μmol·L時,隨著氯磺隆濃度逐漸降低,ALS酶活性逐漸增強,于是選取310 μmol·L的氯磺隆作為檢測ALS酶活性的標準。抗性植株與野生型植株的ALS酶活性檢測結果顯示,在抗性植株中有8株(Chl-3、Chl-5、Chl-8、Chl-11、Chl-12、Chl-18、Chl-20、Chl-22)的ALS酶活性明顯高于野生型的(圖6)。因此,推測這8株定點誘變成功的可能性較大。
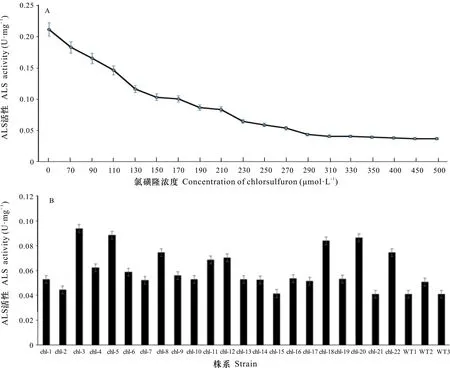
圖 6 氯磺隆對抗性植株和野生型植株的ALS酶活影響Fig. 6 Effects of chlorsulfuron on ALS enzyme activity in resistance and wild-type plants
2.6 抗性煙苗ALS基因突變區序列擴增及測序
利用特異引物,對‘K326’中588位點的保守區及1719位點的保守區進行PCR擴增,產物直接進行測序,擴增產物無雜帶(圖7),在上述22個抗性植株中檢測到2株發生了特定位點的突變,命名為突變株系f11和b18,前者在588位點發生了C→A的突變,后者在1719位點發生了G→T的突變(圖8)。
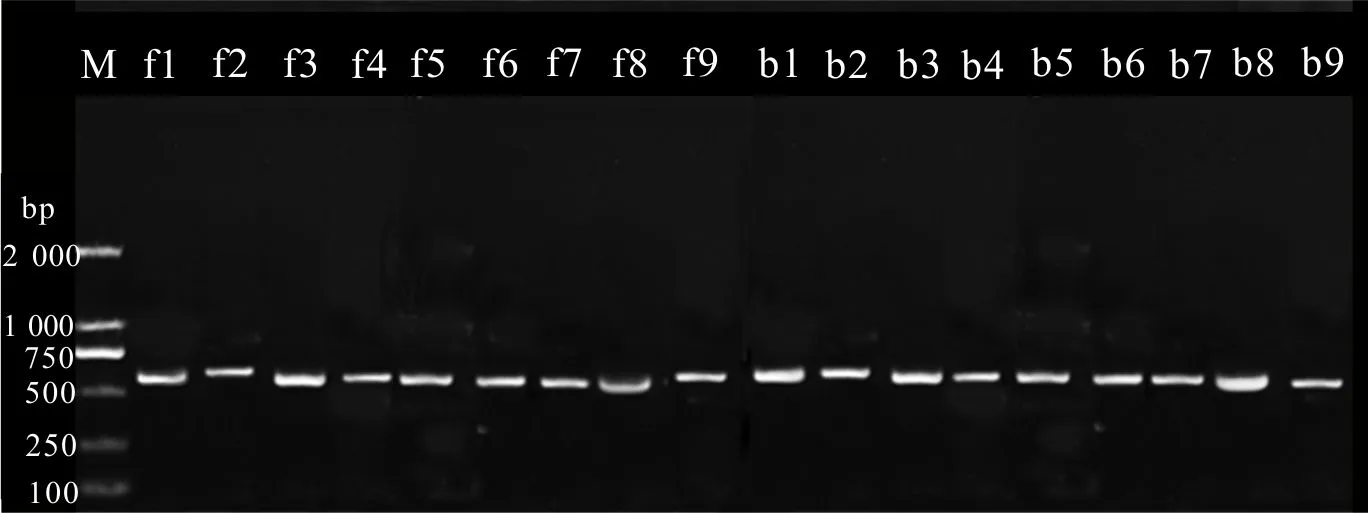
M. DNA 2 000 bp標記; f1-f9. 擴增含588位點區域; b1-b9. 擴增含1719位點區域。M. DNA 2 000 bp Marker; f1-f9. Amplification with segment containing 588 site; b1-b9. Amplification with segment containing 1719 site. 圖 7 含突變位點區段PCR擴增Fig. 7 PCR amplification with segment containing mutation sites
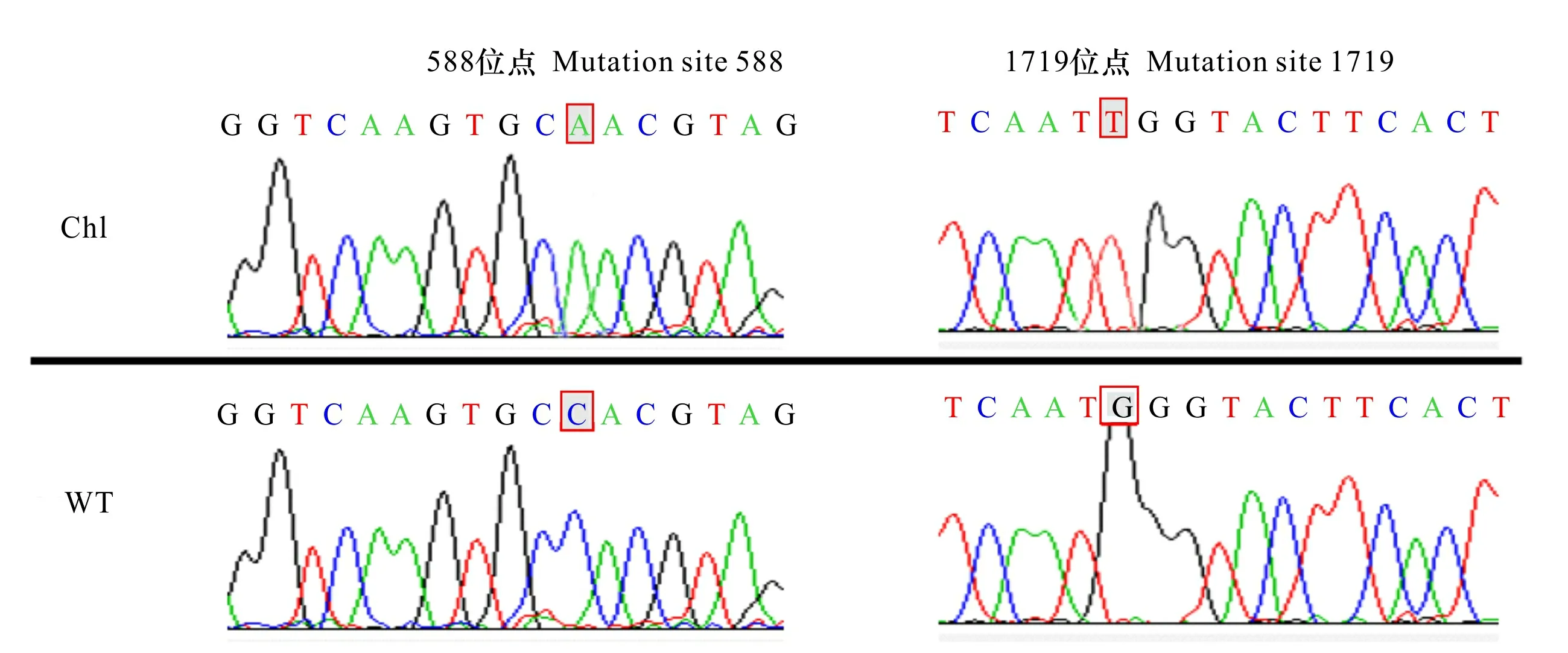
Chl. 抗性植株的測序結果; WT. 野生型的測序結果。Chl. Sequencing result of resistant plants; WT. Sequencing result of wild type plants. 圖 8 含突變位點區段測序Fig. 8 Segment sequencing with mutation site
3 討論與結論
利用生物學手段實現植物體中關鍵代謝基因的定點突變,能提高植物對除草劑的敏感性,從而提高突變植株對除草劑的抗性,但效率較高的基因定點編輯生物學手段CRISPR/Cas及TALEN技術,在操作的過程中引入了外源基因 (Ishino et al., 1987; Michno et al., 2020)。因此,在轉基因作物嚴格管制的今天難以廣泛推廣種植。OMM技術理論上不涉及轉基因,該技術所得新種質,在種植領域上有其得天獨厚的優勢。
煙草品種‘K326’基因測序結果發現,該品種基因序列全部為編碼區,無內含子序列,從起始密碼子至終止子序列完整,而NCBI報道的普通煙草品種與‘K326’品種的基因存在一定差別(Keeler et al., 1993)。NCBI中煙草基因序列長度為2 004 bp,與‘K326’基因序列長度一致;基因序列長度為 1 995 bp,而‘K326’長度為2 010 bp,推測是由品種之間的差異性所致。‘K326’基因與NCBI報道的煙草品種基因盡管序列長度一致,但存在一個堿基的區別,即煙草品種‘K326’基因編碼區序列的第831位發生了A到G的簡并突變,氨基酸密碼子由UUA變成UUG,然而都編碼氨基酸Leu,為簡并突變,經過多次PCR和重復測序證實結果正確無誤,證實這一突變為煙草品種‘K326’本身的差異性。根據‘K326’和‘K326’的保守區,結合Beethan等(1999)、Kochevenko和Willmitzer(2003)的報道,設計了用于‘K326’定點突變的RNA/DNA。
由于Kochevenko和Willmitzer(2003)研究的煙草品種基因并非‘K326’,因此在報道位點上存在位置偏離。為了解決這個問題實驗中找到與其完全配對的目標序列,設計突變位點,命名為Chl-588和Chl-1719。在寡聚核苷酸嵌合體設計過程中,為避免嵌合體的不穩定,對片段進行甲基化保護。Yoon等(1996)、Alexeev等(2000)和Bartlett等(2000)的研究發現,RNA/DNA寡聚核苷酸鏈自身互補形成雙鏈發夾結構,在這種雙鏈結構中,2條互補雙鏈的區別非常明顯,其中一條嵌合體鏈是由5 bp的DNA片段及分別位于該DNA片段兩邊的10 bp的RNA區域組成,而另一條鏈全是由DNA構成,利用載體的特殊結構(兩組胸腺嘧啶脫氧核苷酸連接互補的兩條鏈,在3′末端的“GC”帽子結構及2′-O-甲基RNA殘基),使其在細胞中具有較好的穩定性。因此,實驗設計時采用同樣的方法保證在轉化過程中的穩定性。
本研究利用基因槍轟擊法將定點誘變的RNA/DNA寡聚核苷酸嵌合體Chl-588和Chl-1719轟擊煙草品種‘K326’愈傷組織,定點突變煙草基因,以期得到具有氯磺隆抗性的煙草品種‘K326’植株。耿立召等(2005)提出轟擊距離是基因槍轉化成功的關鍵因素,認為基因槍轟擊煙草的轟擊距離為9 cm。本研究中,探索了基因槍轟擊煙草品種‘K326’愈傷組織的轟擊距離和轟擊壓強,結果得出的轟擊煙草品種‘K326’愈傷組織的最佳距離與蘇寧等(2002)的研究結果一致,這說明在煙草基因槍轟擊實驗中,轟擊條件在不同品種之間差別不大。在氯磺隆篩選過程中,假陽性明顯,分化培養基中篩選出的芽在生根培養時大量死亡,本研究認為可能與愈傷組織在含有氯磺隆的培養基中多次繼代培養造成的,多次繼代培養后使愈傷組織表現了對氯磺隆抗性的假陽性,這一結論與李學寶和毛慧珠(1999)認為的多次繼代培養對抗生素產生抗性基本一致。抗性植株ALS酶活性檢測結果表明,部分抗性植株的ALS酶活性高于野生型,可以初步說明抗性植株目標位點發生了堿基突變與氨基酸發生了突變,具體要看測序的結果。
通過對抗性植株基因測序,目標位點測序結果表明,在目標位點僅有2株抗性植株發生了定點突變,即發生588位點C到A的突變和1719位點由G到T的突變。但是,由于多數抗性植株并未出現定點突變,因此推測實驗結果存在如下可能:(1)煙草染色體為異源四倍體,基因存在2個拷貝(Kochevenko & Willmitzer, 2003),且2個拷貝之間存在一定程度的差異,在基因擴增及RNA/DNA嵌合體定點誘變上帶來一定難度,或是因四倍體在克隆測序時送出的克隆不是突變的那一個拷貝;(2)RNA/DNA嵌合體誘變了其他位點使煙草產生除草劑抗性,煙草AHAS序列的W464位點突變后可以產生抗性,引起植株具有氯磺隆抗性的原因需要進一步去驗證;(3)所得誘變植株假陽性概率較高,成功突變的植株較少,Beethan等(1999)、Kochevenko和Willmitzer (2003)的研究也都報道了相似的結果。目前,OMM法的定點誘變在煙草愈傷的誘變條件還處于初步探索階段。當OMM技術與工程核酸酶TALENs或CRISPR/Cas9一起使用時,觀察到的精確無痕基因組編輯的頻率顯著增加(Sauer et al., 2016)。但是,在編輯的過程中引入外源基因,會使OMM獨特的不涉及轉基因過程的技術優勢蕩然無存,所得作物無法大田推廣。后期,對OMM技術的優化,以及T1、T2代的檢測篩選試驗還在不斷地探索完善過程中。
猜你喜歡
奧秘(創新大賽)(2023年3期)2023-05-06 01:48:20
中國煙草學報(2019年5期)2019-11-14 07:54:12
首都公共衛生(2019年5期)2019-05-21 01:08:34
浙江中西醫結合雜志(2017年2期)2017-01-12 18:23:59
新聞傳播(2016年3期)2016-07-12 12:55:34
當代化工研究(2016年9期)2016-03-20 16:22:08
自動化博覽(2014年6期)2014-02-28 22:32:15
聲屏世界(2014年6期)2014-02-28 15:18:09
西南學林(2013年2期)2013-11-12 12:58:54
中國煙草學報(2012年5期)2012-04-12 06:21:18
