基于對照品和對照提取物的中藥復方質量控制對比研究
2022-09-27 06:31:30周德勇高艷艷林天鳳姜艷艷
世界中醫藥 2022年16期
張 顏 周德勇 王 路 高艷艷 林天鳳 劉 斌 姜艷艷
(北京中醫藥大學中藥學院,北京,102488)
中藥對照提取物是中藥的“標準”提取物,是一類非單體成分對照品,一般通過特殊的提取分離工藝制備而成[1]。具有化學成分組成相對明確、指標成分含量確切、適合多成分分析等特點,可用于薄層色譜或其他色譜鑒別,以及高效液相色譜法的含量測定[2]。對照提取物首次在2005年版《中華人民共和國藥典》中作為中藥標準物質出現,并且隨著藥典修訂工作的不斷推進,更多的對照提取物被收載和應用[3]。2020年版《中華人民共和國藥典·四部》中就收載了人參莖葉總皂苷、三七總皂苷、廣陳皮對照提取物、銀杏葉提取物等23種對照提取物[4]。美國藥典(US Pharmacopoeia,USP)和歐洲藥典(European Pharmacopoeia,EP)對于對照提取物也有相應的收載和應用[5-7]。雖然中藥對照提取物在對照物質總數量中的比例并不大,但是中藥對照提取物具有一些特點和優勢,能夠彌補中藥化學對照品和對照藥材在生產、供應、使用等環節中存在的不足,對中藥質量標準的完善和檢測效率的提高具有促進作用[8]。
中藥復方是中醫用藥的主要形式,在中醫藥醫療活動中發揮著不可替代的作用。中藥復方的功效發揮是其所含有效成分綜合作用的結果,是多成分、多靶點、多途徑作用的體現[9]。因此,中藥復方的質量控制和評價研究應依據其特點開展。近年來,中藥復方質量控制方法研究成為中藥現代化的重要研究領域之一[10]。為了保障中藥復方制劑的安全性與有效性,中醫學者開展了大量的系統研究,旨在建立符合中藥復方特點的系統、科學、全面的質量控制方法。
現階段,中藥質量控制基本都是借鑒化學藥品的控制模式,采用單一對照品評價中藥質量,由于中藥成分的多樣性及復雜性,這種方法往往具有一定的局限性。而對照提取物可以同時對多個組分進行含量測定,體現了中藥整體質量控制的思想。將中藥對照提取物應用于中藥及中藥復方質量控制具有獨特的優勢:相對固定的化學組成,安全、穩定的理化特性;專屬性較強;可以減少化學對照品的使用,降低檢驗成本;配制操作簡單,溶解于有機溶劑后可直接使用,使藥品檢測更加簡便、快捷[11]。因此,開展中藥對照提取物研究并探索將其用于中藥及中藥復方的質量控制具有較大的應用價值,能夠解決目前中藥質量控制研究中的諸多問題。
決明子為豆科植物鈍葉決明CassiaobtusifoliaL.或決明(小決明)CassiatoraL.的干燥成熟種子。具有清熱明目、潤腸通便的功效。臨床常用于目赤澀痛、羞明多淚、頭痛眩暈、目暗不明、大便秘結[12]。決明子作為脂康顆粒中的重要藥味,由決明子、枸杞子、桑椹、紅花、山楂四味中藥組成,具有滋陰清肝、活血通絡的作用,臨床上可用于治療肝腎陰虛挾瘀所引起的高脂血癥等疾病[12]。課題組前期系統開展了決明子萘并吡喃酮類對照提取物制備及定殖研究,并將其應用于決明子藥材的質量控制與評價[13-17]。文獻及前期研究結果顯示,將中藥對照提取物用于中藥及中藥復方的質量控制,穩定性、重復性良好,結果準確度高、可行性強[18-21]。在此基礎上,以脂康顆粒為例,分別采用化學對照品法和對照提取物法,建立含有決明子的中藥制劑脂康顆粒的含量測定方法,并對測定結果進行對比分析。進一步探討決明子萘并吡喃酮類對照提取物用于中藥復方質量控制中的可行性和適用性,為全面評價決明子藥材及復方質量提供研究基礎,為中藥對照提取物在中藥復方質量控制與評價工作中的適用性和可行性提供科學依據。
1 材料與方法
1.1 材料
1.1.1 分析樣品 枸杞子、桑葚、山楂、紅花4種藥材和各批次脂康顆粒(批號:170310)樣品均由同仁堂(望京店)提供。
1.1.2 試劑 決明子苷B2對照品、決明子苷C2對照品、紅鐮霉素-6-O-β-D-龍膽二糖苷對照品、決明子苷C對照品由北京中醫藥大學中藥學院自制,經過結構鑒定及色譜檢測計算,純度均大于98%;乙腈、甲醇為色譜純(賽默飛世爾科技(中國)有限公司,美國,批號:155805);純凈水;甲醇為分析純(天津市致遠化學試劑有限公司,批號:2020121025);乙醇為分析純(天津市大茂化學試劑廠,批號:20190806);乙酸為分析純(汕頭市西隴化工廠有限公司,批號:080312)。
1.1.3 儀器 循環水式多用真空泵(上海振捷實驗設備有限公司,型號:SHB-Ⅲ);數控超聲波清洗器(昆山市超聲儀器有限公司,型號:KQ-500DE);高效液相色譜儀(沃特世科技(上海)有限公司,美國,型號:1525型);Agilent ZORBAX SB-C18色譜柱(4.6 mm×250 mm,5 μm)(安捷倫科技(中國)有限公司,美國,型號:ZORBAX SB-C18);十萬分之一電子分析天平(北京賽多利斯儀器有限公司,型號:Sartorious BT 25S)。
1.2 方法
1.2.1 對照品用于脂康顆粒含量測定
1.2.1.1 高效液相色譜(High Performance Liquid Chromatography,HPLC)分析 色譜柱:Agilent ZORBAX SB-C18色譜柱(250 mm×4.6 mm,5 μm);流動相:甲醇乙腈(5∶4)混合溶液(A)-1%乙酸水(B)等度洗脫(0~30 min,19%A);流速:1 mL/min;檢測波長:278 nm;柱溫:35 ℃。
1.2.1.2 對照品溶液的制備 精密稱取決明子苷B2對照品2.75 mg、決明子苷C2對照品2.35 mg、紅鐮霉素-6-O-β-D-龍膽二糖苷對照品4.73 mg、決明子苷C對照品7.96 mg,置于50 mL量瓶中,加甲醇溶解并稀釋至刻度,搖勻,即得混合對照品溶液A。精密吸取3 mL混合對照品溶液A,置于10 mL量瓶中,加甲醇稀釋至刻度,得混合對照品溶液B;精密吸取3 mL混合對照品溶液B,置于10 mL量瓶中,加甲醇稀釋至刻度,得混合對照品溶液C;重復以上步驟直至制得混合對照品溶液E。
1.2.1.3 供試品溶液的制備 精密稱定脂康顆粒1 g,加入50 mL水溶解,上樣于內徑為2 cm,高度為25 cm的大孔吸附樹脂柱,柱體積為80 mL,先用320 mL 20%乙醇洗脫除雜后,再用480 mL 50%乙醇洗脫,收集洗脫液,減壓回收溶劑至干燥,殘渣加30%甲醇溶解,轉移至25 mL量瓶中,加30%甲醇并稀釋至刻度,搖勻,即得供試品溶液。
1.2.1.4 陰性樣品溶液的制備 取枸杞子、桑葚、山楂、紅花4味藥材適量,按照處方3∶3∶3∶1的比例,加水煎煮2次,第1次1.5 h,第2次1 h,濾過,濾液合并,減壓濃縮成相對密度為1.05~1.10(60 ℃)的浸膏,加入麥芽糊精適量,混勻,噴霧干燥;浸膏粉中加入麥芽糊精適量和硬脂酸鎂5 g,混勻,干法制粒,即得陰性樣品。按供試品溶液的制備方法制得陰性樣品溶液。
1.2.1.5 線性關系考察 精密吸取“1.2.1.2”項下制得的混合對照品溶液A~F各10 μL,注入液相色譜儀,測定色譜峰峰面積。以對照品進樣量為橫坐標(X),色譜峰峰面積為縱坐標(Y),進行線性回歸。
1.2.1.6 儀器精密度試驗 分別精密稱取同一供試品溶液6份,每次進樣10 μL,以色譜峰峰面積,計算相對標準偏差(Relative Standard Deviation,RSD)。
1.2.1.7 中間精密度試驗 分別精密稱取同一批脂康顆粒6份,每份1 g,按“1.2.1.3”項下方法由不同的分析人員在不同的時間制備脂康顆粒樣品溶液,分別采用Waters 1525-2998-2707和Waters 1525-2489高效液相色譜儀測定峰面積。
1.2.1.8 供試品溶液穩定性試驗 取“1.2.1.3”項下供試品溶液,分別于制備后0 h、2 h、4 h、8 h、16 h、24 h進樣,測定色譜峰峰面積。
1.2.1.9 重復性試驗 分別精密稱取同一批脂康顆粒6份,每份1 g,按“1.2.1.3”項下方法制備供試品溶液,分別進樣,測定峰面積。
1.2.1.10 回收率試驗 分別精密稱取同一批脂康顆粒6份,每份0.5 g,加入對照品溶液適量,按“1.2.1.3”項下方法制備加樣回收供試品溶液,分別精密吸取10 μL,注入液相色譜儀,測定色譜峰峰面積。
1.2.2 對照提取物用于脂康顆粒含量測定
1.2.2.1 HPCL分析 色譜條件與對照品色譜條件相同。
1.2.2.2 對照提取物溶液的制備 精密稱取已標定含量的決明子萘并吡喃酮類對照提取物12.88 mg,置于25 mL容量瓶中,加30%甲醇超聲溶解,靜置,定容,搖勻,即得對照提取物溶液A。精密吸取3 mL對照提取物溶液A于10 mL量瓶中,加甲醇稀釋至刻度,得對照提取物溶液B;精密吸取3 mL對照提取物溶液B于10 mL量瓶中,加甲醇稀釋至刻度,得對照提取物溶液C;重復以上步驟直至制得對照提取物溶液E。
1.2.2.3 供試品溶液的制備 精密稱定脂康顆粒1 g,加入50 mL水溶解,上樣于內徑為2 cm,高度為25 cm的大孔吸附樹脂,柱體積為80 mL,320 mL 20%乙醇洗脫除雜后,用480 mL 50%乙醇洗脫,收集洗脫液,減壓回收溶劑至干燥,殘渣加30%甲醇溶解,轉移至25 mL量瓶中,加30%甲醇稀釋至刻度,搖勻,即得供試品溶液。
1.2.2.4 陰性樣品溶液的制備 取枸杞子、桑葚、山楂、紅花4味藥材適量,按照處方3∶3∶3∶1的比例,加水煎煮2次,第1次1.5 h,第2次1 h,濾過,濾液合并,減壓濃縮成相對密度為1.05~1.10(60 ℃)的浸膏,加入麥芽糊精適量,混勻,噴霧干燥;浸膏粉中加入麥芽糊精適量和硬脂酸鎂5 g,混勻,干法制粒,即得陰性樣品。按“1.2.1.3”項下方法制得陰性樣品溶液。
1.2.2.5 線性關系考察 精密吸取“1.2.2.2”項下混合對照提取物溶液A~F各10 μL,注入液相色譜儀,測定色譜峰峰面積。以對照品進樣量為橫坐標(X),色譜峰峰面積為縱坐標(Y),進行線性回歸。
1.2.2.6 儀器精密度試驗 分別精密稱取同一供試品溶液6份,每次進樣10 μL,以色譜峰峰面積,計算RSD。
1.2.2.7 中間精密度試驗 分別精密稱取同一批脂康顆粒6份,每份1 g,按“2.2.3”項下方法由不同的分析人員在不同的時間制備脂康顆粒樣品溶液,分別采用Waters 1525-2998-2707和Waters 1525-2489高效液相色譜儀測定峰面積。
1.2.2.8 供試品溶液穩定性試驗 取“1.2.2.3”項下供試品溶液,分別于制備后0 h、2 h、4 h、8 h、16 h、24 h進樣,測定4種指標性成分的色譜峰峰面積。
1.2.2.9 重復性試驗 分別精密稱取同一批脂康顆粒6份,每份1 g,按“1.2.2.3”項下方法制備供試品溶液,分別進樣,測定峰面積。
1.2.2.10 回收率試驗 分別精密稱取同一批脂康顆粒6份,每份0.5 g,加入對照提取物溶液適量,按“1.2.2.3”項下方法制備供試品溶液,分別精密吸取10 μL,注入液相色譜儀,測定色譜峰峰面積。
1.2.3 復方脂康顆粒含量測定 精密稱取12批脂康顆粒粉末,按“1.2.2.3”項下方法制備供試品溶液,分別以4個對照品和已知含量的決明子對照提取物為對照,采用高效液相色譜法,測定4種成分的含量,并對不同方法所得的結果進行t檢驗。
2 結果
2.1 對照品用于脂康顆粒含量測定結果
2.1.1 HPLC分析 混合對照品、供試品和陰性樣品色譜圖見圖1。
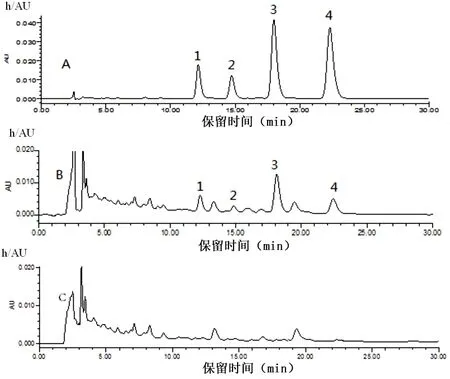
圖1 混合對照品、脂康顆粒樣品及陰性樣品的HPLC色譜圖
2.1.2 線性關系 決明子苷B2線性范圍為0.004 5~0.550 0 μg;決明子苷C2線性范圍為0.003 8~0.470 0 μg;紅鐮霉素-6-O-β-D-龍膽二糖苷線性范圍為0.007 7~0.946 0 μg決明子苷C線性范圍為0.012 9~1.592 0 μg。
2.1.3 儀器精密度 RSD值分別為0.58%、0.87%、0.90%、0.79%,均小于3%,表明儀器精密度良好。
2.1.4 中間精密度 決明子苷B2、決明子苷C2、紅鐮霉素-6-O-β-D-龍膽二糖苷及決明子苷C的含量分別為0.08%(RSD=0.77%),0.010%(RSD=1.20%),0.15%(RSD=0.93%),0.04%(RSD=1.05%),表明該方法中間精密度良好。
2.1.5 供試品溶液穩定性 RSD值分別為1.01%,0.72%,1.23%和0.57%。表明該溶液在制備后24 h內穩定性良好。
2.1.6 重復性 RSD值均小于3%,表明重復性良好。
2.1.7 回收率 RSD值均小于3%,表明回收率良好。見表1。
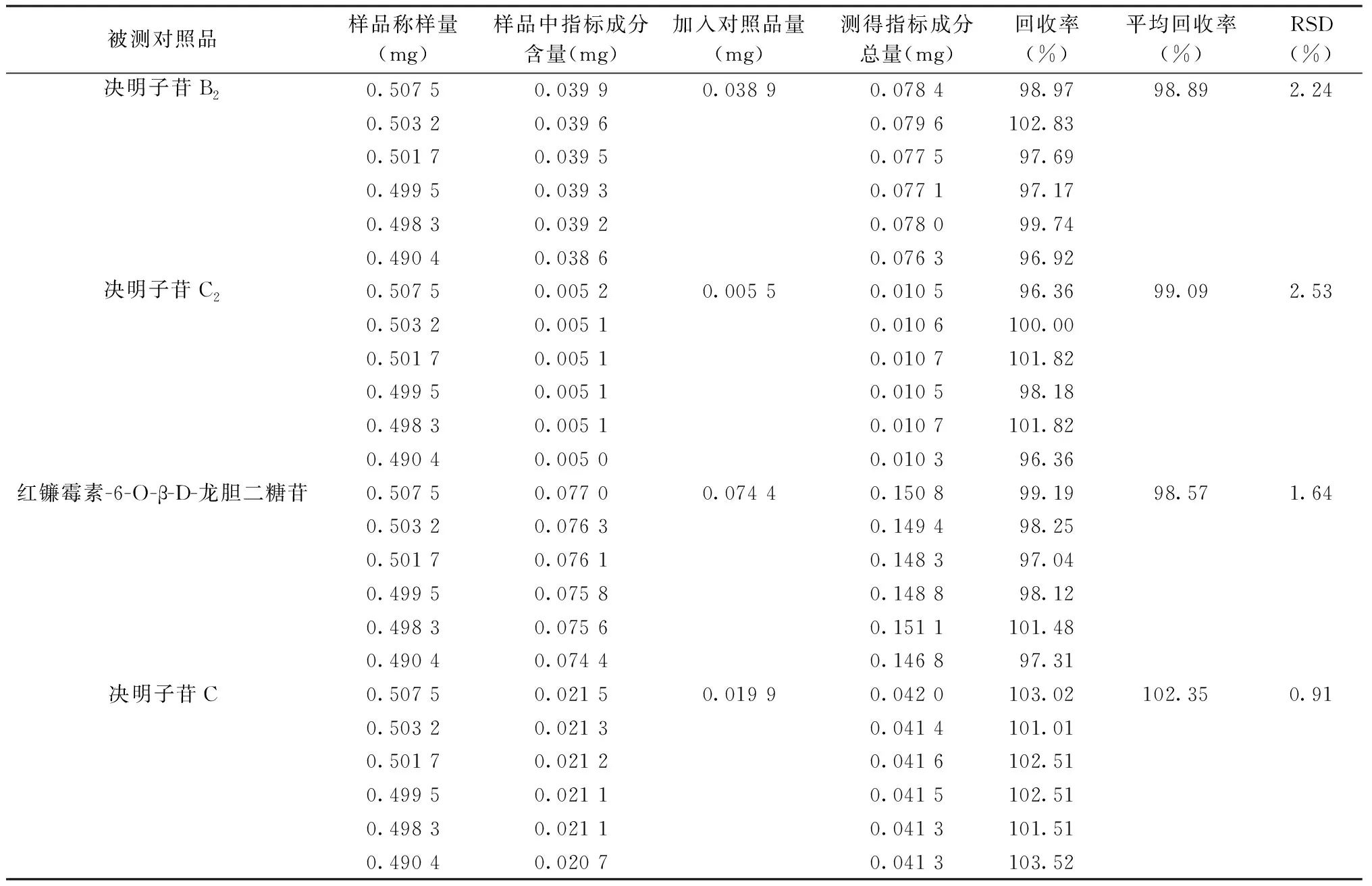
表1 加樣回收率考察結果(n=6)
2.2 對照提取物用于脂康顆粒含量測定
2.2.1 HPLC分析 對照提取物、供試品及陰性樣品溶液色譜圖見圖2。
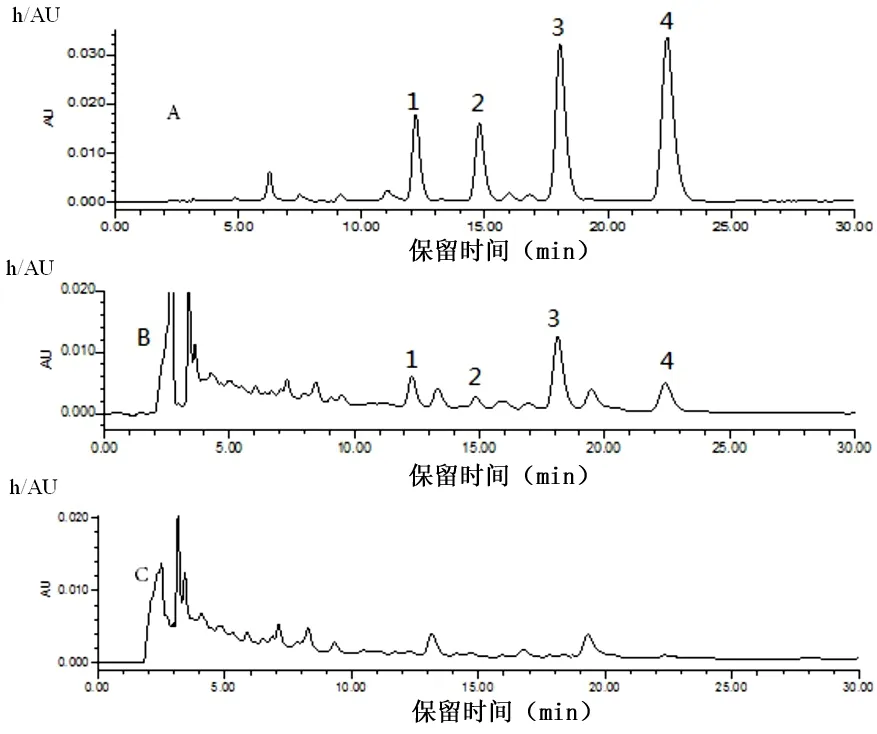
圖2 對照提取物、脂康顆粒樣品及陰性樣品的HPLC色譜圖
注:A.對照提取物;B.供試品;C.陰性樣品;1.決明子苷B2;2.決明子苷C2;3.紅鐮霉素-6-O-β-D-龍膽二糖苷4.決明子苷C
2.2.2 線性關系 決明子苷B2線性范圍為0.004 5~0.550 0 μg;決明子苷C2線性范圍為0.003 8~0.470 0 μg;紅鐮霉素-6-O-β-D-龍膽二糖苷線性范圍為0.007 7~0.946 0 μg決明子苷C線性范圍為0.012 9~1.592 0 μg。
2.2.3 儀器精密度 RSD值分別為0.74%、0.91%、0.85%、1.20%,均小于3%,表明儀器精密度良好。
2.2.4 中間精密度 決明子苷B2、決明子苷C2、紅鐮霉素-6-O-β-D-龍膽二糖苷及決明子苷C的含量分別為0.08%(RSD=0.91%),0.01%(RSD=1.23%),0.151%(RSD=0.95%),0.04%(RSD=1.02%),表明該方法中間精密度良好。
2.2.5 供試品溶液穩定性 RSD值分別為1.10%,1.43%,0.97%和1.24%。表明該溶液在制備后24 h內穩定性良好。
2.2.6 重復性 RSD值均小于3%,表明該方法重復性良好。
2.2.7 回收率 RSD值均小于3%,表明回收率良好。見表2。
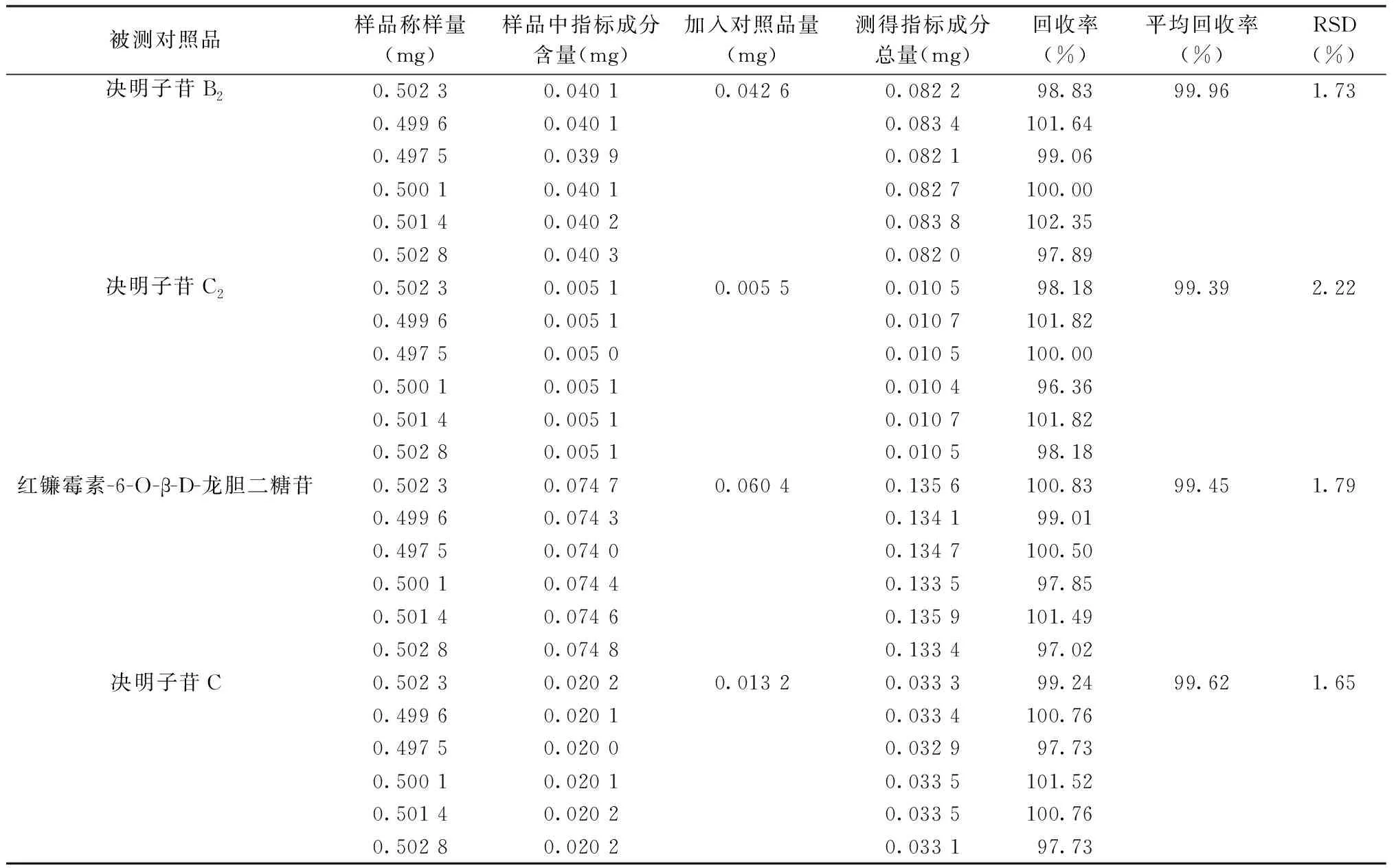
表2 加樣回收率考察結果(n=6)
2.3 復方脂康顆粒含量測定 決明子苷B2、決明子苷C2、紅鐮霉素-6-O-β-D-龍膽二糖苷和決明子苷C的P值分別為0.233、0.082、0.192、0.198,P值均大于0.05,說明對照提取物可以作為脂康顆粒質量控制中的對照物質,代替相應的化學對照品進行質量評價。見表3。
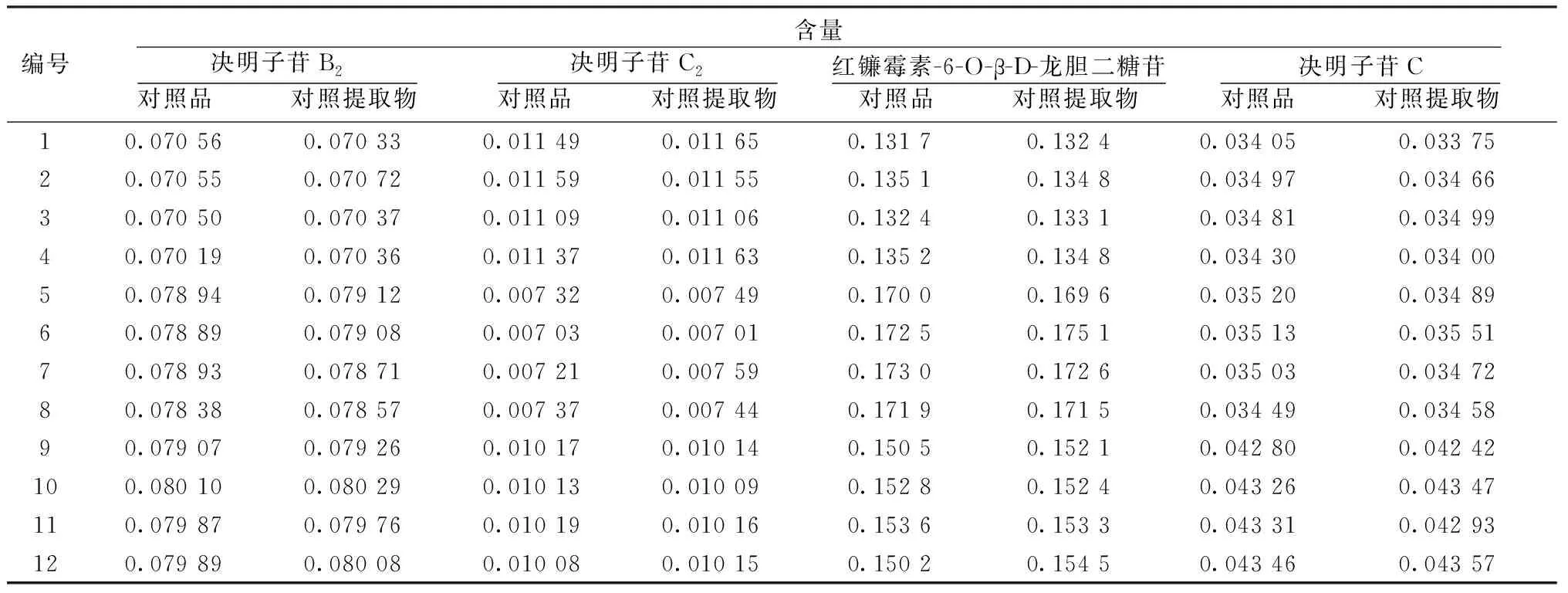
表3 12批脂康顆粒含量測定(%,n=3)
3 討論
中藥是在傳統中醫藥理論指導下用于防治疾病的藥物,其質量直接影響中醫臨床用藥的安全性和有效性。一方面,隨著中藥市場的擴大、中藥品種及產地多元化,一些傳統的質量評價方法已經遠遠不能滿足實際需要;另一方面,隨著科學技術的高速發展,中醫藥在國際上的地位和影響力不斷增強,因此為了更好地實現中藥現代化和國際化,中藥標準物質及質量控制也正向著多指標成分同步分析的方向發展[18]。而中藥對照提取物正是順應了這一發展趨勢,在中藥整體的質量控制中體現出了巨大的優越性,為實現多成分測定的質量控制新模式做出了貢獻。
本研究中首次建立了基于決明子萘并吡喃酮類對照提取物(主成分為決明子苷B2、決明子苷C2、紅鐮霉素-6-O-β-D-龍膽二糖苷及決明子苷C)的中藥復方脂康顆粒含量測定方法,采用配對樣本t檢驗法分析比較對照提取物和對照品測定結果的異同,發現二者的結果不存在顯著差異,表明對照提取物法與對照品法所得結果一樣準確可靠,決明子對照提取物可以作為脂康顆粒質量控制中的對照物質,代替相應的化學對照品實現對中藥復方的質量控制和評價。進一步明確了對照提取物在決明子復方質量控制中的適用性和可行性,為中藥對照提取物應用于中藥復方質量控制提供了參考。
猜你喜歡
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中老年保健(2021年4期)2021-12-01 11:19:40
中老年保健(2021年4期)2021-08-22 07:08:32
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
金橋(2020年7期)2020-08-13 03:07:00
基層中醫藥(2020年12期)2020-07-22 06:34:38
中國生殖健康(2019年2期)2019-08-23 08:12:08
基層中醫藥(2018年6期)2018-08-29 01:20:20
產品可靠性報告(2017年7期)2017-09-05 09:49:12
汽車觀察(2016年3期)2016-02-28 13:16:26
