ADH1B基因甲基化對卵巢癌細胞增殖及凋亡的影響
2022-09-21 06:34:16季維雪李澤蓮
安徽醫科大學學報 2022年8期
季維雪,孫 磊, 江 玉, 陳 潁, 李澤蓮, 李 敏, 肖 蘭
作者單位:1安徽醫科大學附屬阜陽醫院婦產科,阜陽 2361122安徽醫科大學第一附屬醫院婦產科,合肥 230022
卵巢癌是女性生殖系統常見三大惡性腫瘤之一,病死率居首位,化療耐藥是導致卵巢癌療效不佳的重要因素之一[1]。越來越多研究[2]顯示卵巢癌發生、發展及耐藥與表觀遺傳學改變密切相關。表觀遺傳修飾包括DNA甲基化、組蛋白修飾等,其中DNA甲基化異常在卵巢癌耐藥形成中起著重要作用。文獻[3]報道,通過DNA甲基化抑制劑逆轉 DNA甲基化狀態,為靶向治療卵巢癌提供了一個新的靶點。酒精脫氫酶(alcohol dehydrogenase, ADH)是一種多態性酶,乙醇脫氫酶1B (alcohol dehydrogenase 1B, ADH1B)也稱為ADH2,目前ADH1B與癌癥的聯系主要集中在酒精代謝和飲酒行為等方面[4]。新近研究[5]表明甲基化下調的ADH2與乳腺癌不良預后相關,人腫瘤細胞中ADH2基因亦受表觀遺傳機制調控,提示ADH2基因有望成為潛在腫瘤個體化治療靶點。該研究擬體外觀察5-Aza-dc對卵巢癌細胞增殖和凋亡的影響,旨在探討ADH1B基因甲基化與卵巢癌發生發展的關系。
1 材料與方法
1.1 材料與試劑細胞株:卵巢癌細胞C13K及OV2008為本室保存;組織樣本選取2019年1月—2021年5月就診于醫院婦產科且均經病理學診斷卵巢癌患者55例,FIGO分期:Ⅰ~Ⅱ期18例,Ⅲ~Ⅳ期37例。同期因子宮肌瘤需手術患者21例(留取正常卵巢上皮)。卵巢癌組患者的平均年齡(50.4±9.6)歲,對照組平均年齡(44.6±7.3)歲。排除自身免疫病及其他系統腫瘤,術前均未接受新輔助化療、免疫及靶向治療,經手術切除的新鮮組織標本均在30 min內。研究對象均簽署知情同意書,并通過醫院倫理委員會審查。DNA純化試劑盒購自德國Qiagen公司;去甲基化制劑5-Aza-dc購自美國Sigma公司;PRMI Medium 1640培養基、小牛血清均購自美國Gibco公司;ADH1B兔多克隆抗體購自英國Abcam公司;Hoechst 33258染色液購自上海碧云天公司;Trizol試劑購自美國Invitrogen公司;逆轉錄試劑盒及SYBR Green Master Mix購自日本TaKaRa公司;CCK-8檢測試劑盒購自上海碧云天公司。
1.2 方法
1.2.1生物信息學分析 生物信息學分析網站GEPIA(http://gepia.cancer-pku.cn/)分析ADH1B在卵巢癌的表達,數據來源于癌癥和腫瘤基因圖譜(cancer genome atlas,TCGA)。
1.2.2細胞培養及干預分組 C13K和OV2008細胞于含15%小牛血清1640培養液,置37℃、5% CO2培養箱培養。當細胞融合度達(70~80)%時,0.25%胰蛋白酶消化傳代,取對數生長期細胞進行實驗。5-Aza-dc設低、中及高3個濃度組,分別為0.5、2.5及10 μmol/L,分別干預48 h。
1.2.3去甲基化藥物對卵巢癌細胞毒性作用 各組細胞按1×104細胞/孔濃度種至96孔板,繼續培養24 h。細胞增殖:5-Aza-dc (0.5、2.5及10 μmol/L)干預48 h,加10 μl CCK-8溶液,繼續培養4 h,450 nm波長處各孔吸光度值。實驗均重復3次,取平均值。
1.2.4Hoechst33258染色法觀察細胞形態變化 兩株細胞以2×105細胞/孔濃度接種于6孔培養板,細胞貼壁后加高濃度5-Aza-dc(10 μmol/L)繼續培養48 h,PBS洗滌細胞2次,4%多聚甲醛固定30 min,PBS洗滌細胞2次,Hoechst 33258(5 mg /L)在室溫下染色10 min后棄去染色液,PBS洗2遍,熒光顯微鏡下觀察,拍照。正常細胞核Hoechst著色形態呈圓形,淡藍色;凋亡細胞核濃集固縮而呈亮藍色。
1.2.5甲基化特異性PCR (methylation specific PCR, MSP) 采用DNA甲基化修飾試劑盒對基因組DNA進行甲基化修飾,具體操作按試劑盒說明書進行。PCR反應條件:95 ℃預變性12 min;94 ℃變性30 s,退火溫度58 ℃,72 ℃延伸45 s,45個循環;72 ℃延伸10 min。擴增后產物用2%瓊脂糖凝膠電泳,凝膠成像系統觀察分析。ADH1B MSP甲基化引物序列F:5′-CCAGGGATTAGGAGTGGACC-3′,R: 5′-GGAGGGGAAGAGCAGTTGTC-3′; 未甲基化引物序列, UF: 5′-CAGTGTGGAAAATGCAGAG-3′; UR:5′-GTGACCTTGGCAACGTTA-3′。MSP結果判定:僅擴增出甲基化條帶者為完全甲基化;同時出現甲基化條帶和非甲基化條帶者為部分甲基化; 僅出現非甲基化條帶者為未甲基化。
1.2.6qRT-PCR檢測ADH1BmRNA水平 收集組織標本及5-Aza-dc(0.5、10 μmol/L)作用兩組細胞,將組織樣本置液氮中研磨成勻漿,TRIzol法提取組織和細胞RNA,反轉錄合成cDNA。PCR引物序列,ADH1B,F: 5′-GTGGCACAAGCGTCATCGTAGG-3′,R: 5′-TTCCAGGTGCGTCCAGTCAGTAG-3′;β-actin,F: 5′-AGAAGGCTGGGGCTCATTTG-3′,R: 5′-AGGGGCCATCCACAGTCTTC-3′。2-△△Ct法計算各組細胞中ADH1B基因mRNA相對表達量,實驗重復3次。
1.2.7免疫印跡檢測ADH1B蛋白水平 30 μg蛋白質樣品行SDS-PAGE電泳,濕轉至硝酸纖維膜上;5% BSA室溫封閉2 h,0.05% Tween20的TBS緩沖液(TBST)漂洗,10 min×3次;加入ADH1B(1 ∶1 000)一抗,b-actin一抗(1 ∶5 000),4 ℃過夜,1×TBST漂洗3遍,對應辣根過氧化物酶標記二抗(1 ∶5 000),37 ℃搖床溫育2 h,ECL顯色曝光,采集照片。
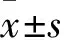
2 結果
2.1 ADH1B在卵巢癌中表達被抑制為明確ADH1B在OC中表達情況,根據TCGA數據中ADH1B在426例OC患者樣本和88例正常樣本表達的差異結果顯示,ADH1B在腫瘤樣本中比正常樣本低,差異有統計學意義(P<0.05) , 見圖1。
2.2 組織及兩株細胞中ADH1B基因表達qRT-PCR證實,正常卵巢上皮組織中ADH1B表達水平為(6.72±1.41),高于卵巢癌組織(1.15±0.67),差異有統計學意義(t=-5.742,P<0.01);OV2008細胞ADH1B基因表達為C13K細胞5.82倍,差異有統計學意義(t=-7.313,P<0.01)。

圖1 ADH1B 在卵巢癌及正常卵巢上皮組織中相對表達量
2.3 5-Aza-dc對細胞存活率比較低濃度5-Aza-dc(0.5 μmol/L)對兩株卵巢癌細胞生長無明顯抑制作用(P>0.05);中濃度5-Aza-dc(2.5 μmol/L)對兩株卵巢癌細胞生長有一定抑制作用(t=9.234,P<0.01;t=15.416,P<0.01),當5-Aza-dc增至高濃度10μmol/L時,兩株細胞生長抑制差異顯著(F=40.082,P<0.001;F=80.384,P<0.001)(表1)。

表1 各組細胞存活率比較
2.4 細胞凋亡形態學的變化熒光顯微鏡下,經高濃度5-Aza-dc(10 μmol/L)作用48 h后,OV2008及C13K細胞均出現典型細胞凋亡形態學改變,表現為核染色質濃縮、碎裂,而未加藥兩株對照細胞核均呈均勻藍色熒光,見圖2。

圖2 兩株細胞細胞凋亡Hoechst 33258熒光染色 ×200
2.5 5-Aza-dC對ADH1B啟動子甲基化影響結果顯示:ADH1B基因啟動子區在OV2008及SKOV3細胞株中呈完全甲基化;高濃度5-Aza-dC (10 μmol/L)處理48 h后,可見ADH1B基因啟動子區出現U帶,M帶未完全消失,提示ADH1B啟動子區甲基化被部分逆轉,見圖3。

圖3 5-Aza-dC處理前后兩株細胞ADH1B基因啟動子甲基化
2.6 5-Aza-dC對ADH1B mRNA表達影響OV2008細胞中ADH1B mRNA表達高于C13K細胞;5-Aza-dC(0.5、10 μmol/L)作用48 h后,OV2008及C13K細胞ADH1B mRNA均有所升高,尤以高濃度5-Aza-dC(10 μmol/L)處理后OV2008及C13K細胞ADH1B mRNA升高差異顯著(F=8.093,P<0.01;F=21.928,P<0.01),圖4。

圖4 細胞中ADH1B mRNA表達水平比較
2.7 免疫印跡檢測ADH1B蛋白水平結果顯示,無5-Aza-dC作用,OV2008及C13K細胞中ADH1B蛋白呈較低表達狀態,且C13K中表達較之OV2008更低;低濃度5-Aza-dC(0.5 μmol/L)未引起OV2008及C13K細胞中ADH1B蛋白改變,而高濃度5-Aza-dC(10 μmol/L)使OV2008及C13K細胞中ADH1B蛋白均表達增加,見圖5A。ADH1B/b-actin對蛋白圖像掃描半定量統計結果表明,低濃度5-Aza-dC(0.5μmol/L)處理后,兩株細胞ADH1B蛋白表達無明顯改變(P>0.05);高濃度5-Aza-dC (10 μmol/L)處理使ADH1B蛋白表達較兩株對照組及低濃度組升高,差異有顯著性(F=12.059,P<0.01;F=30.384,P<0.01),見圖5B。

圖5 各組細胞ADH1B蛋白表達
3 討論
ADH1B基因編碼 I 類ADH的β亞基,定位于染色體4q23。Gharpure et al[6]發現ADH1B異常表達使卵巢癌細胞分泌金屬基質蛋白酶7、白細胞分化抗原26和組織蛋白酶從而促進腫瘤進展。ADH1B在鼻咽癌細胞中表達下調,鼻咽癌細胞過表達ADH1B后可抑制鼻咽癌細胞的增殖和遷移能力,逆轉細胞的惡性程度[7]。另有研究[8]指出ADH1B可能為卵巢癌細胞化療耐藥的候選基因之一。本研究qRT-PCR結果及卵巢癌TCGA數據分析均提示ADH1B在OC患者樣本中低表達,據此課題組推測ADH1B基因可能作為抑癌基因參與了卵巢癌的發生發展,但其具體調控機制目前尚未明確,有必要進行深入探討。
腫瘤細胞DNA異常甲基化是一種常見表觀遺傳學改變,這種異常修飾可抑制基因轉錄,使抑癌基因喪失功能[9-10]。基因啟動子區DNA甲基化是導致基因失活的重要原因之一,而基因啟動子甲基化的潛在可逆性為新型抗癌藥物的開發提供了機會,這些藥物可重新激活沉默的腫瘤抑制基因[11]。以5-Aza-dc為代表的類核苷甲基轉移酶抑制劑,通過與DNA甲基轉移酶共價結合抑制其活性,降低基因甲基化水平,已廣泛應用于逆轉腫瘤細胞的異常甲基化,誘導因甲基化而沉默的抑癌基因重新表達,抑制腫瘤細胞生長,從而達到殺傷腫瘤細胞的效應[12-13]。為探討ADH1B基因甲基化與卵巢癌發生發展的關系,本研究將 5-Aza-dC作用于兩株卵巢癌細胞株,通過CCK-8 法檢測5-Aza-dc對人卵巢癌細胞生長的影響,結果表明,5-Aza-dc對兩株卵巢癌細胞增殖均有抑制作用,且該作用與 5-Aza-dc濃度呈正相關;同時,Hoechst 33258熒光染色結果顯示,高濃度5-Aza-dc處理后,細胞核染色質濃縮或碎裂,出現典型細胞凋亡形態,表明5-Aza-dc誘導卵巢癌細胞發生凋亡;RT-PCR結果顯示,ADH1B mRNA表達與啟動子區甲基化程度密切相關,在完全甲基化兩株卵巢癌細胞中,ADH1B mRNA表達較低,而5-Aza-dC作用48 h后,兩株細胞ADH1B甲基化均被部分逆轉,ADH1B在mRNA和蛋白水平表達均顯著提高,該結果提示5-Aza-dC可能通過沉默DNA甲基轉移酶而降低抑癌基因ADH1B 啟動子區域的甲基化水平誘導其重新表達。
綜上所述,ADH1B基因甲基化在OC發生發展中發揮重要作用,5-Aza-dc可部分逆轉卵巢癌細胞啟動子區ADH1B甲基化,誘導ADH1B基因重新表達,抑制腫瘤生長,促進細胞凋亡,從而發揮抗腫瘤效應。本研究也為將5-Aza-dc應用于卵巢癌臨床治療提供一定的理論依據。
