雙金屬有機骨架衍生的Fe-CrSe/C負極材料制備及儲鋰性能
2022-09-16 09:29:02陳修棟簡佳琴劉培芳曹小華劉金杭
無機化學學報 2022年9期
陳修棟 簡佳琴 嚴 平 劉培芳 曹小華 劉金杭*,
(1九江學院化學化工學院,九江 332005)(2北京化工大學有機無機復合材料國家重點實驗室,北京 100029)(3信陽師范學院分析測試中心,信陽 464000)(4江西省生態化工工程技術研究中心,九江 332005)
人類社會經濟快速發展,造成了許多環境問題以及燃料供應的枯竭,當前發展清潔能源至關重要[1]。由于水能、風能、太陽能、潮汐能等清潔能源具有間歇性的特征,對儲能器件提出了更高的要求[2]。目前許多高效的儲能器件已經被開發,諸如鋰離子電池、鋰金屬電池、鈉離子電池、鋰硫電池和超級電容器等[3]。鋰離子電池以其充電速度快、能量密度高、無記憶效應和循環壽命長等優勢受到了廣泛的關注[4]。但是商業化的石墨負極理論比容量只有372 mAh·g-1且倍率性能較差[5-6],不能滿足未來高儲能器件的需求,故具有優異電化學性能的負極材料得到了廣泛關注。
具有孔隙率高、比表面積高、熱穩定性好、密度低和晶體結構有序等優點的金屬有機骨架(MOFs)是由金屬離子與有機配體通過配位鍵連接而成的多孔材料[7-8]。然而,MOFs直接作為電極材料時,往往具有低電導率和較差的循環穩定性[9]。MOFs可以被用作犧牲模板來獲得各種多孔納米金屬衍生材料及其復合材料(金屬氧化物、金屬硫化物、金屬硒化物),其中金屬硒化物具有更高的初始庫侖效率和更穩定的循環性能。由于MOFs中的有機配體在惰性氣氛中退火碳化,可使得由MOFs衍生的功能材料具有多孔以及空心微粒結構,有利于鋰離子的運輸。同時,碳化得到的碳框架封裝了電化學活性的納米粒子,提高了電極材料的電子導電性,確保鋰離子和電子的快速擴散。因此,合成原位碳包覆金屬硒化物作為鋰離子電池負極材料將會得到廣泛應用。
我們以對苯二甲酸(H2DBC)和三氯化鉻(CrCl3)為原料,通過微波法合成Cr-MOF,后續通過加入九水合硝酸鐵繼續進行微波反應以得到雙金屬有機骨架材料Fe-Cr-MOF。Fe-Cr-MOF和Cr-MOF分別與硒粉混合在氮氣氣氛下進行退火合成Fe-CrSe/C和CrSe/C衍生材料,并分別作為鋰離子電池負極組裝成半電池進行測試。相比之下,Fe-CrSe/C展現出優良的電化學性能(在100 mA·g-1的電流密度下循環150圈還能維持891.6 mAh·g-1的比容量),具有潛在的應用前景。
1 實驗部分
1.1 試劑與儀器
H2DBC、CrCl3、九水合硝酸鐵、聚偏二氟乙烯(PVDF)、N,N-二甲基甲酰胺(DMF)、1-甲基-2-吡咯烷酮(NMP)、無水乙醇均為分析純(AR)且均購自國藥集團化學試劑有限公司。乙炔黑、鋰片、銅箔、電解液、聚丙烯多孔膜(隔膜)均購自蘇州乾民化學試劑有限公司。
所用儀器:單模微波反應器,上海屹堯儀器科技發展有限公司;SmartLab型X射線粉末衍射儀(XRD,CuKα輻射,波長λ=0.154 06 nm,管電壓為40 kV,管電流為30 mA,掃描范圍為5°≤2θ≤85°,掃描速度為 5(°)·min-1),日本理學公司;Hitachi S-4800型場發射掃描電子顯微鏡(SEM,工作電壓1 kV),日本日立公司;Tecnai G2 F20 S-TWIN型多功能場發射透射電子顯微鏡(TEM,工作電壓20 kV),美國FEI公司;激光拉曼光譜測試儀,英國Renishaw公司;ASAP 2460型比表面積和孔徑分布測試儀(BET),美國麥克儀器公司;STA449F3 Jupiter型熱重(TG)分析儀,德國耐馳公司。
1.2 材料制備
稱取0.5 mmol CrCl3和0.5 mmol H2DBC分別溶于5 mL DMF得到A和B溶液,然后將B緩慢加入A中攪拌均勻,在200℃微波反應30 min。隨后稱取0.5 mmol九水合硝酸鐵,直接加入上述反應體系中,接著在150℃下微波反應30 min[8-9]。用DMF、乙醇、水洗滌上述反應生成物,除去其中未反應的離子,然后置于真空干燥箱中干燥得到雙金屬有機骨架前體材料(Fe-Cr-MOF)。將Fe-Cr-MOF與硒粉按照質量比4∶1混合均勻后放置于瓷舟中,在氮氣保護下,500℃退火處理2 h得到復合電極材料(Fe-CrSe/C)。上述操作中不加入九水合硝酸鐵且不進行后續微波反應,則最終得到的MOF前體材料為Cr-MOF。Cr-MOF以上述步驟相同的條件進行硒化得到CrSe/C。
1.3 電池的組裝
按照6∶2∶2的質量比分別稱取活性材料、PVDF、乙炔黑,置于小試管內,加入溶劑NMP,再用勻漿機在35 000 rad·min-1條件下均漿6次(每次1 min),將其混合均勻。將均勻的材料涂在銅箔上,隨后放入真空干燥箱內,在60℃條件下烘干12 h制成電極片,其每個極片上活性物質為1.4 mg·cm-2。鋰離子半電池的組裝是在充滿氬氣的手套箱中進行,將所制電極片用作工作電極,金屬鋰片作為對電極,Celgard 2300聚丙烯作為隔膜,1 mol·L-1LiPF6溶液作為電解液,組裝成CR2032型紐扣電池。
1.4 電化學性能測試
電化學測試均在室溫條件下進行,電池的恒電流充放電測試是通過深圳新威爾測試系統進行,電壓范圍為0.005~3.0 V。同時,使用上海辰華的CH1760E型電化學工作站對電池進行交流阻抗測試和循環伏安(CV)測試,CV掃描速率為0.1 mV·s-1,其中交流阻抗的測試范圍為0.01 Hz~100 kHz。
2 結果與討論
2.1 材料的結構和形貌分析
圖1a和1b分別為Fe-Cr-MOF和Cr-MOF的氮氣吸附-脫附等溫線及孔徑分布圖。從圖中可知,Fe-Cr-MOF和Cr-MOF材料孔徑分布都呈現出介孔結構特征,但Fe-Cr-MOF(1 220 m2·g-1)的比表面積遠大于Cr-MOF(48 m2·g-1)。這表明,與Cr-MOF相比,Fe-Cr-MOF為電解液浸潤提供了更多的有效面積,同時也為鋰離子在電極材料中的運輸提供更多通道。圖1c為Cr-MOF和Fe-Cr-MOF的TG分析曲線,由圖可知,這2種材料質量的損失都隨溫度升高而增加。Cr-MOF在100℃左右質量開始出現減小,而Fe-Cr-MOF在200℃左右質量才出現減少,二者的質量損失都源于本身的熱分解。Cr-MOF的質量損失明顯要大于Fe-Cr-MOF的,這表明Fe-Cr-MOF比Cr-MOF的熱穩定性更好。
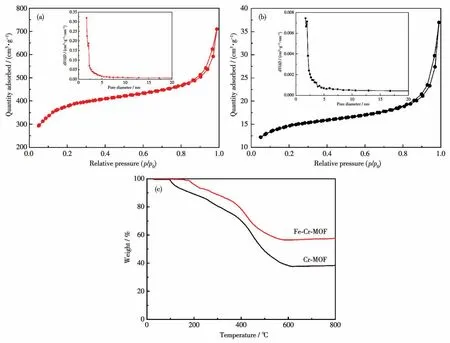
圖1 (a)Fe-Cr-MOF和(b)Cr-MOF的氮氣吸附-脫附等溫線;(c)Cr-MOF和Fe-Cr-MOF的TG曲線Fig.1 Nitrogen adsorption-desorption isotherms of(a)Fe-Cr-MOF and(b)Cr-MOF;(c)TG curves of Cr-MOF and Fe-Cr-MOF
圖2a為CrSe/C和Fe-CrSe/C的拉曼譜圖,其中,在1 330和1 550 cm-1附近出現了2個特征峰,分別歸屬于碳的D峰和G峰,二者的相對強度比(ID/IG)表示無定型碳或缺陷碳在復合材料中所占比例。從圖中可看出CrSe/C和Fe-CrSe/C的ID/IG值分別為1.02和1.08,因此,Fe-CrSe/C比CrSe/C具有更多的無定形碳。圖2b為CrSe/C和Fe-CrSe/C的XRD圖,從圖中可知,Fe-CrSe/C在2θ=41.8°和54.8°處的峰分別歸屬為CrSe的(102)和(103)晶面。另外,Fe-CrSe/C在2θ=64.9°處有一個較強的衍射峰,主要歸屬于FeCr合金的(200)晶面。CrSe/C在2θ=27.6°、60.1°和69.1°處的峰分別主要歸屬于C峰和CrSe的(201)、(202)晶面。Fe-CrSe/C和CrSe/C的強峰位置不同,主要歸因于Fe離子的引入造成了峰位置的變化[10]。
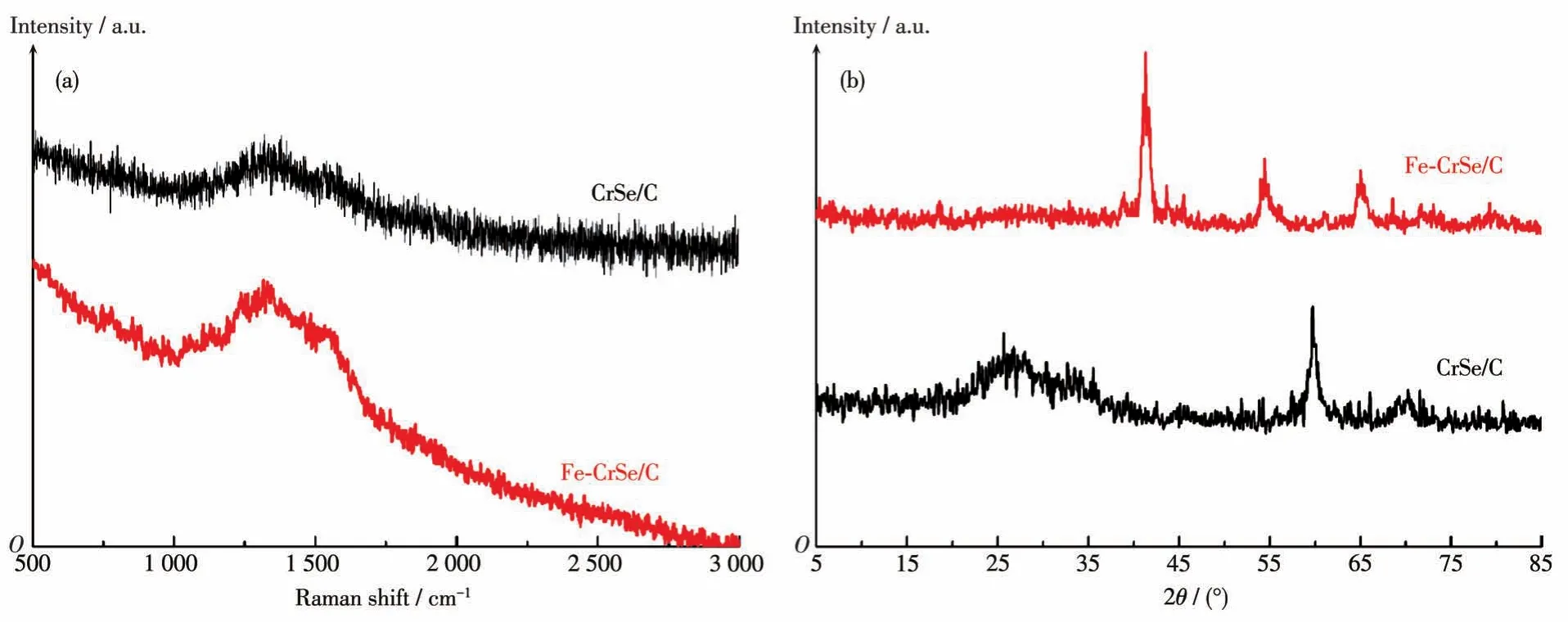
圖2 CrSe/C和Fe-CrSe/C的拉曼譜圖(a)和XRD圖(b)Fig.2 Raman spectra(a)and XRD patterns(b)of CrSe/C and Fe-CrSe/C
圖3a和3c分別為CrSe/C的SEM圖和TEM圖,圖3b和3d分別為Fe-CrSe/C的SEM圖和TEM圖,從圖中可觀察出CrSe/C和Fe-CrSe/C均形成了納米顆粒狀的形貌。元素映射顯示Fe-CrSe/C中的C、Se、Cr和Fe均勻分布,進一步佐證了雙金屬有機骨架衍生材料的成功合成(圖3e~3i)。

圖3 (a)CrSe/C和(b)Fe-CrSe/C的SEM圖;(c)CrSe/C和(d)Fe-CrSe/C的TEM圖;(e~i)Fe-CrSe/C的元素映射Fig.3 SEM images of(a)CrSe/C and(b)Fe-CrSe/C;TEM images of(c)CrSe/C and(d)Fe-CrSe/C;(e-i)Element mappings of Fe-CrSe/C
2.2 電化學性能分析
Fe-CrSe/C的CV曲線如圖4a所示,在首圈陰極掃描過程中,Fe-CrSe/C分別在0.8、1.4和2.0 V附近出現了一個較強的還原峰,這些峰主要可以歸因于金屬鋰與Fe-CrSe/C發生反應以及固體電解質界面膜的形成。在陽極掃描過程中,Fe-CrSe/C分別在1.8和2.3 V附近出現了一個強氧化峰,這主要由于脫鋰反應。從第2圈循環開始,可以觀察到還原峰向1.1 V位置移動,峰重疊性好,這說明Fe-CrSe/C電極具有優良的可逆性。圖4b展示了Fe-CrSe/C的前3圈充放電曲線,該曲線與CV曲線相匹配,Fe-CrSe/C首圈初始放電和充電容量分別為1 229.7和958.4 mAh·g-1,首圈庫侖效率為77.93%。首圈庫侖效率低,主要是首圈循環過程中電極表面的固體電解質界面膜的生成和電解液分解引起的。在100 mA·g-1電流密度下測試了CrSe/C和Fe-CrSe/C的循環性能,如圖4c所示。CrSe/C電極在150圈循環后只有254 mAh·g-1的可逆比容量。相比之下,Fe-CrSe/C在150圈循環后仍保持了891.6 mAh·g-1的較高充電容量。另外,還進行了CrSe/C和Fe-CrSe/C首圈循環后的電化學阻抗譜(EIS)測試(圖4d),所測譜圖由高頻區半圓與低頻區的直線組成,半圓直徑越小,對應的界面電荷轉移阻抗越小,更有利于電化學反應。從圖中可知,Fe-CrSe/C(144 Ω)相比于CrSe/C(169 Ω)具有更小的電荷轉移阻抗,表明Fe-CrSe/C電極具有更好的電導率,進一步解釋了Fe-CrSe/C比CrSe/C具有更優異電化學性能。
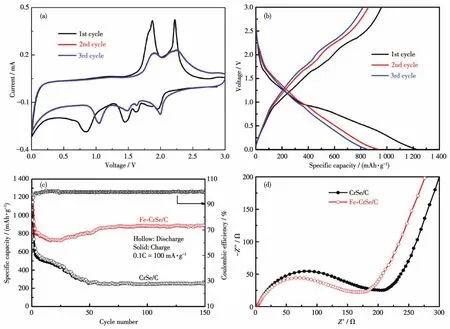
圖4 Fe-CrSe/C的CV曲線(a)和充放電曲線(b);CrSe/C和Fe-CrSe/C的循環曲線圖(c)和循環首圈后的EIS譜圖(d)Fig.4 CV curves(a)and charge-discharge curves(b)of Fe-CrSe/C;Cycle curves(c)and EIS spectra after the first cycle(d)of CrSe/C and Fe-CrSe/C
如圖5a所示,測試了Fe-CrSe/C在不同掃描速率下的CV曲線。在不同掃描速率下的CV曲線形狀相似,這說明Fe-CrSe/C電極具有優良的循環可逆性[11]。將lgi相對于lgv作圖,其中i和v分別代表電流和掃描速率,則可獲得斜率b(圖5b)。眾所周知,如果b值接近0.5,則擴散貢獻在電極反應中起著關鍵作用,而當b接近1.0時,則是電容貢獻主導了反應過程。在2個典型峰的位置,b值分別為0.28和0.36,這表明Fe-CrSe/C在充放電過程中主要是以擴散貢獻為主[12-13]。此外,根據相應公式計算電容貢獻與擴散貢獻[14],如圖5c所示。在不同的掃描速率下計算的電容貢獻和擴散貢獻所占的比例如圖5d所示。在0.1 mV·s-1時,擴散貢獻占90.94%。將掃描速率提高到 0.2、0.3、0.4和0.5 mV·s-1之后,擴散貢獻分別為87.64%、85.28%、83.38%和81.78%。在掃描速率的不斷提高下,電容貢獻逐漸上升,這是因為鋰離子在低掃描速率下相對更容易擴散,但是在較高的掃描速率時擴散則會被抑制。同時,Fe-CrSe/C在高掃描速率下的較高電容貢獻清楚地展示出其獨特的納米微粒結構為鋰離子提供了豐富的活性位點,從而提高了循環的穩定性。
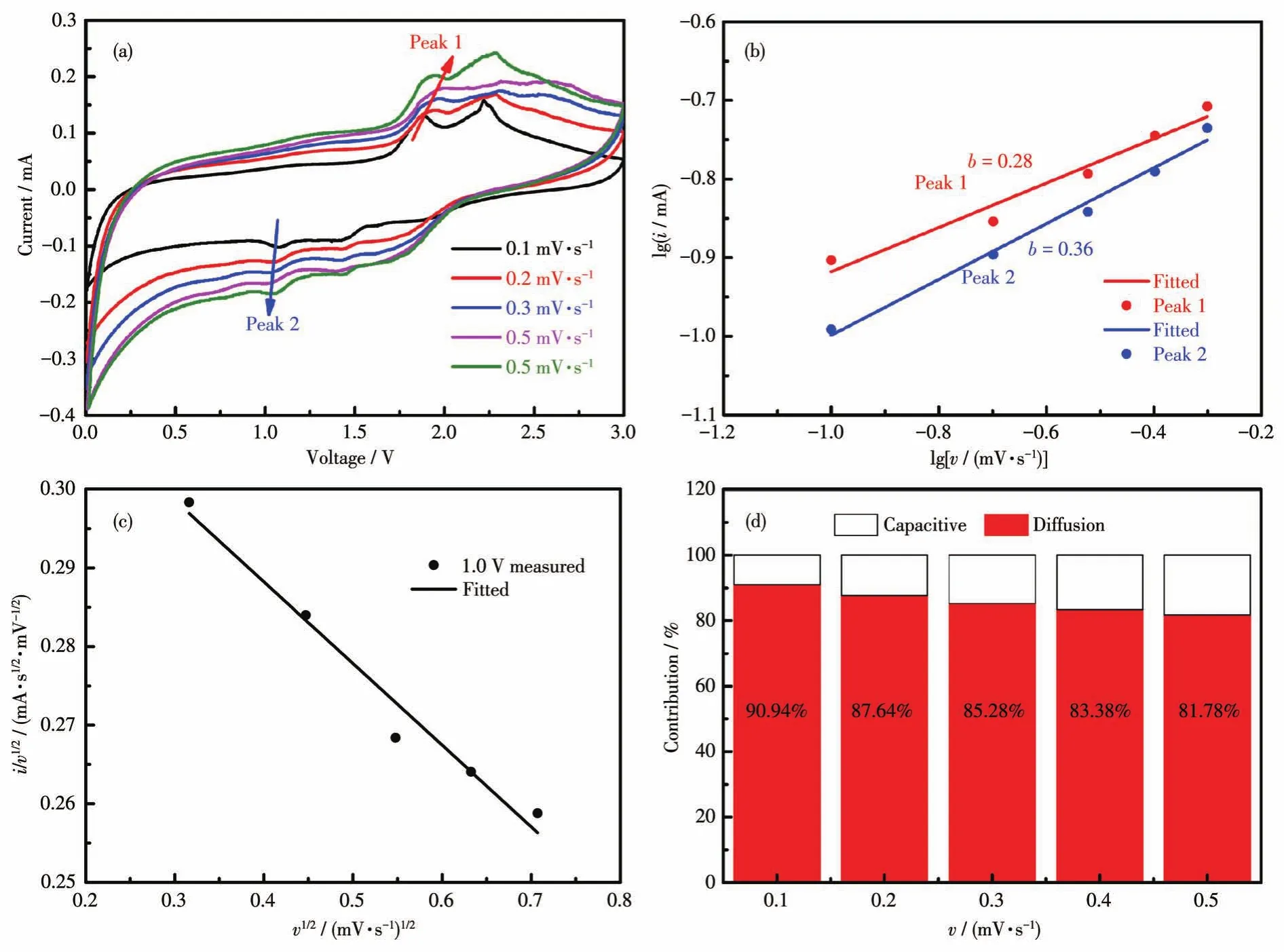
圖5 (a)Fe-CrSe/C在不同掃描速率下的CV曲線圖;(b)lg i vs lg v圖;(c)v1/2vs i/v1/2圖;(d)在不同掃描速率下電容貢獻和擴散貢獻所占的比例Fig.5 (a)CV curves of Fe-CrSe/C at different scan rates;(b)lg i vs lg v plots;(c)v1/2vs i/v1/2plot;(d)Proportion of capacitance contribution and diffusion contribution at different sweep rates
在首圈充放電過程中在不同電壓條件下進行了Fe-CrSe/C的原位EIS測試。如圖6所示,電荷轉移阻抗在整個過程中逐漸降低,這表明了電極Fe-CrSe/C的活化和高可逆性,進一步證明了Fe-CrSe/C具有較好的電化學性能。
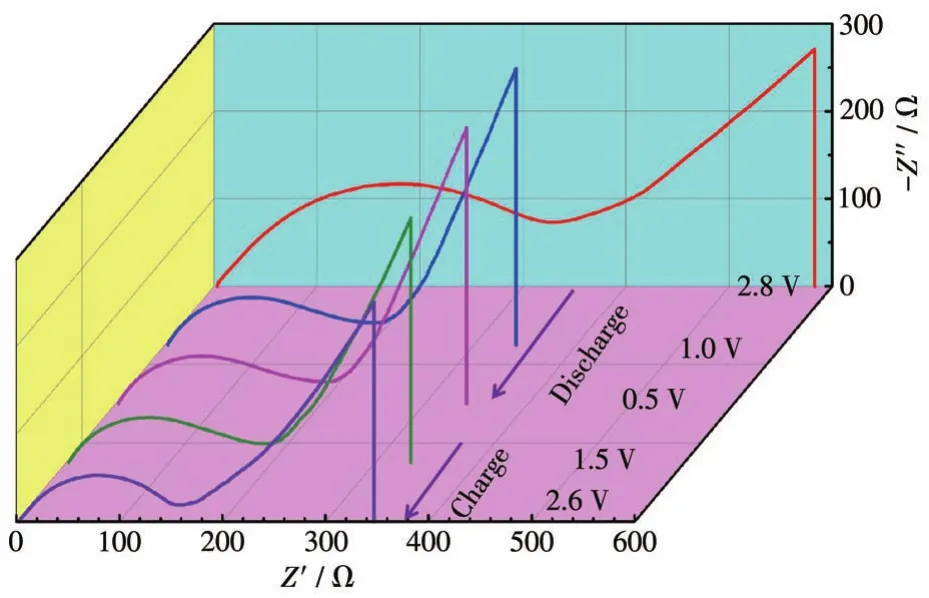
圖6 在不同充放電電壓下Fe-CrSe/C的原位EIS譜圖Fig.6 In situ EIS spectra of Fe-CrSe/C at different charge and discharge voltages
3 結 論
通過微波法合成了2種新型MOF衍生材料CrSe/C和Fe-CrSe/C,將其作為鋰離子電池負極材料,組裝成半電池進行電化學性能測試。結果表明,雙金屬有機骨架衍生的硒化物(Fe-CrSe/C)具有較好的電化學性能。在電流密度100 mA·g-1下,Fe-CrSe/C材料在循環150圈后比容量還能保持891.6 mAh·g-1。Fe-CrSe/C電極材料的研究為將來開發高性能的鉻基MOF衍生電極材料提供了理論基礎。
