表面硫化和磷化提升鉬酸鎳的析氫性能
2022-09-16 09:28:58姜仁政常峻華張金鳳李孟江謝英鵬
無機化學學報 2022年9期
關鍵詞:催化劑
姜仁政 常峻華 高 穎 張金鳳 李孟江 謝英鵬*,
(1沈陽化工大學化學工程學院,沈陽 110142)(2沈陽化工大學材料科學與工程學院,沈陽 110142)
0 引 言
氫能具有能量密度大(120~142 MJ·kg-1)、熱值高、轉化效率高等特點,是一種理想的能源載體,也被視為21世紀最具發展潛力的清潔能源[1-3]。目前H2的主要來源是通過化石燃料制取的“灰氫”,生產過程伴隨著大量二氧化碳(CO2)排放。利用可再生能源產生的電能驅動水分解反應產生H2,不僅可以獲得“綠氫”(過程無CO2排放),還可以解決可再生能源的存儲和運輸問題,是最理想的能源利用方式[4-5]。電解水析氫反應(HER)需要引入合適的催化劑來降低反應過電勢,提高析氫效率。貴金屬Pt是最高效的HER催化劑,然而Pt的稀缺和價格昂貴限制了電解水制氫技術的大規模應用[6-7]。因此,開發儲量豐富、經濟高效的電解水催化劑是生產“綠氫”的關鍵,同時也面臨著巨大挑戰。
過渡金屬Mo/Ni基硫化物或磷化物因具有優異的電催化性能,成為有望替代貴金屬的HER催化劑。研究發現,通過構建異質結構,并結合摻雜、形貌調控和缺陷工程等方法,可以顯著提高Mo/Ni基化合物的HER性能。例如,Mo摻雜Ni3S2/NixPy形成豐富的摻雜異質結構,通過協同優化OH*、O*和OOH*中間物種的生成吉布斯自由能,加快了電化學分解水反應動力學速率[8]。泡沫鎳(NF)負載MoS2/Ni3S2具有高活性異質界面,利于電荷傳遞,提升HER和析氧反應(OER)動力學和反應活性[9]。高分散Mo原子摻雜具有豐富氧缺陷結構的Ni2P/NiMoO4-y,提供了更多的HER協同活性位點,提高了導電性,促進了傳質過程[10]。3D分級異質結構MoP/Ni2P自支撐電極,因其結構特點以及磷化物組分間的協同作用,擁有優越的分解水性能[11]。此外,相比于晶相材料,非晶相電催化劑材料(如非晶相納米片結構CoMoS[12])具有短程有序、長程無序的特點,其較大的比表面積可以為HER過程提供大量的開放活性位點。
Mo/Ni基氧化物,如鉬酸鎳(NiMoO4),也是被廣泛研究的HER催化劑。雖然NiMoO4自身HER性能有限,但通過如前所述的各種改性方法也可大幅提高其電催化活性。例如,在前期研究中我們發現,P摻雜和磷酸鹽負載的NiMoO4具有優異的HER性能[13],這主要是因為P摻雜提高了NiMoO4的導電性,而表面負載的磷酸鹽提供了豐富的表面反應位點。
我們首先制備了NiMoO4納米陣列自支撐泡沫鎳電極(NMO/NF),再通過水熱硫化法在其表面形成非晶相硫化物殼層,最后通過氣相磷化法進行表面處理獲得硫化物和磷酸鹽共同修飾的PS-NMO/NF電極。由于具有非晶相硫化物和磷酸鹽,PS-NMO/NF電極的表面活性位大幅增加,而且硫化物與磷酸鹽之間形成的異質界面利于電子傳輸,從而大幅提高電極的分解水析氫性能。結果表明,在1 mol·L-1KOH電解液中,電流密度為10和100 mA·cm-2時,PS-NMO/NF所對應的析氫過電勢分別為93和180 mV,并展現出優異的長時穩定性。
1 實驗部分
1.1 原料與試劑
NF導電基體購自昆山佳勝電子有限公司,厚度為1.5 mm,每平方厘米孔數為17個;四水合鉬酸銨((NH4)6Mo7O24·4H2O)、六水合硝酸鎳(Ni(NO3)2·6H2O)、水合次亞磷酸鈉(NaH2PO2·H2O)、氫氧化鉀(KOH)、硫代乙酰胺(CH3CSNH2)均為分析純,購自國藥控股化學試劑有限公司;鹽酸(HCl)、丙酮(CH3COCH3)、無水乙醇(CH3CH2OH)均為分析純,購自天津大茂化學試劑有限公司;去離子水(18.2 MΩ·cm)采用德國Millipore系統制備。
1.2 電催化劑的制備
PS-NMO/NF制備過程如圖1所示,首先通過水熱過程制備NMO/NF,再通過二次水熱硫化過程制備硫化改性的NMO/NF(S-NMO/NF),最后通過磷化過程獲得PS-NMO/NF。具體步驟如下:
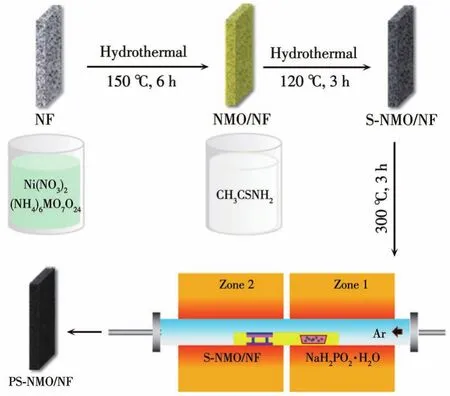
圖1 PS-NMO/NF的制備流程示意圖Fig.1 Schematic diagram of the preparation process of PS-NMO/NF
制備NMO/NF:按照前期工作中的合成方法[13],首先配制15 mL(NH4)6Mo7O24(15 mmol·L-1)和Ni(NO3)2·6H2O(35 mmol·L-1)的水溶液;隨后將上述均一溶液轉入20 mL的聚四氟乙烯反應釜內膽中,并將預處理的NF(3 mol·L-1HCl超聲清洗30 min,然后分別用丙酮、乙醇和去離子水超聲清洗3次)斜靠于內膽壁;將內膽裝入反應釜密封后,在烘箱中150℃下反應6 h。反應完成后取出NF電極,經去離子水和無水乙醇多次清洗,60℃干燥過夜后得到NMO/NF電極。此外,反應釜內膽中生成的粉末樣品同樣經過清洗和干燥,獲得NMO樣品。
制備S-NMO/NF:將NMO/NF電極置于15 mL硫代乙酰胺(2.6 mmol·L-1)的水溶液中進行二次水熱處理。在烘箱中120℃下反應3 h,經去離子水和無水乙醇多次清洗,60℃干燥過夜后得到S-NMO/NF電極。將NMO粉末經過同樣水熱處理過程,經清洗干燥后獲得S-NMO粉末樣品。
制備PS-NMO/NF:將S-NMO/NF置于高溫管式爐下游,上游放置2.0 g NaH2PO2·H2O粉末,在Ar氣氛中(流量為40 mL·min-1)300℃(升溫速率為3℃·min-1)熱處理3 h,得到PS-NMO/NF電極,PS-NMO負載量約為7.2 mg·cm-2。將S-NMO粉末經過同樣磷化處理,獲得PS-NMO粉末。
1.3 電催化劑的表征方法
用日本理學的D/max-2500 PC型X射線衍射儀(XRD)分析物相組成,輻射源為 CuKα1射線,λ=0.154 18 nm,管電壓為50 kV,管電流為200 mA,掃描 步幅為 0.02°,2θ=10°~80°。用德 國 Zeiss的SUPRA 55 SAPPHIRE場發射掃描電子顯微鏡(SEM)表征電極的微觀形貌,用配置的牛津OXFORD X射線電制冷能譜儀(EDS)分析元素組成和分布,加速電壓為5~30 kV。用高分辨透射電鏡(HRTEM,TECNAI G20)表征電催化劑的微觀結構和晶格結構,測試用粉體樣品是通過超聲剝離所制備電極獲得。用美國Thermo的ESCALAB 250型X射線光電子能譜(XPS)分析樣品表面的元素組成和價態,輻射源為AlKα射線(1 486.6 eV),所有鍵能采用污染碳C1s軌道鍵能(284.6 eV)進行校準。
1.4 電化學實驗
采用單槽三電極體系,以自制的自支撐電極為工作電極、石墨棒為對電極、Ag/AgCl電極為參比電極,在1.0 mol·L-1KOH電解液中進行電化學實驗。利用CHI 660E電化學工作站進行數據采集。所有電勢采用可逆氫電極電勢(RHE)進行校準(ERHE=EAg/AgCl+0.204 6+0.059 1pH),電流密度基于電極的反應幾何面積(1 cm2)進行計算。在自動IR補償(90%)模式下,掃描速率控制在2 mV·s-1,采用線性掃描伏安法(LSV)測得HER極化曲線。然后,根據Tafel方程(η=blg|j|+a,其中η是過電位,b是Tafel斜率,j是電流密度)和線性擬合獲得Tafel斜率。采用不同掃描速率下的循環伏安(CV)曲線測得雙電層電容(Cdl)和電化學活性表面積(ECSA)。利用電化學阻抗譜(EIS)技術在析氫過電位100 mV下測定電荷傳遞阻力,掃描范圍為100 kHz~0.01 Hz,振幅為5 mV。運用恒電流技術考察電催化劑的穩定性。
2 結果與討論
2.1 電催化劑的表征
采用XRD分析電催化劑的物相組成和晶體結構。由于自支撐電極材料中基體Ni骨架含量較多,造成鉬酸鎳及其表面生長的活性組分特征峰較弱,難以分析。因此,采用NMO、S-NMO和PS-NMO粉末進行XRD分析,結果見圖2。由圖可知,位于2θ=14°、26°、27°和34°處的衍射峰可歸屬于β-NiMoO4的特征峰(PDF No.12-0348),在29°出現的微弱衍射峰屬于α-NiMoO4(PDF No.33-0948)。而在 2θ=10°(插圖)、21°、22°、27°、30°和 31°的衍射峰對應 NiMoO4·H2O(PDF No.13-0128)[14-15]。通過比較3個樣品的XRD圖可以看出,NMO主要由β-NiMoO4和NiMoO4·H2O兩相構成,同時含有極少量的α-NiMoO4。依次經液相硫化和氣相磷化處理后,得到的S-NMO和PS-NMO粉體仍然以混合相NiMoO4為主,未發現硫化物和含磷化合物的特征峰存在,這表明硫化、磷化作用僅能在NiMoO4表層生成含量很少的硫化物和磷化物。
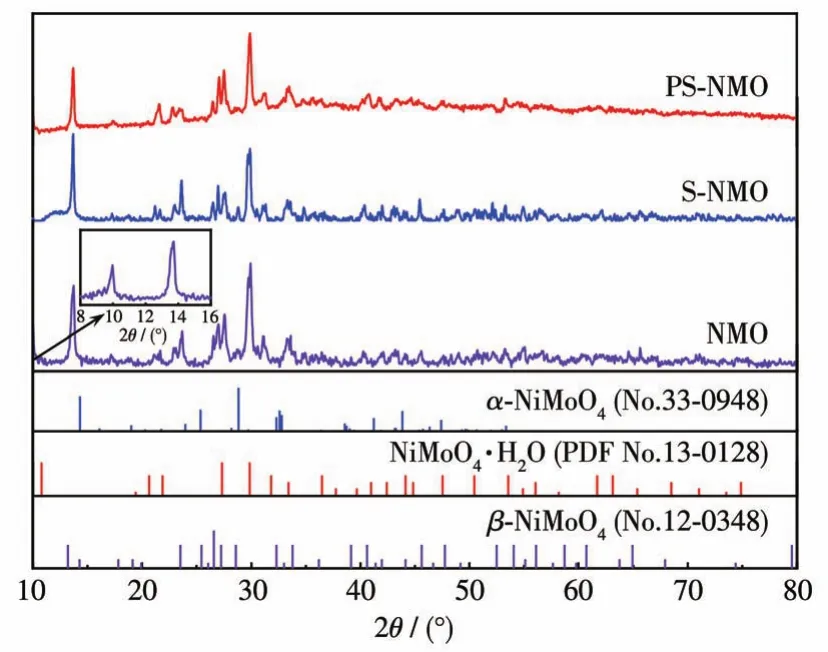
圖2 NMO、S-NMO和PS-NMO的XRD圖Fig.2 XRD patterns of NMO,S-NMO,and PS-NMO
圖3a為NMO/NF的SEM照片,由圖可見,在NMO/NF中約5 μm長的NMO納米棒以團簇形式生長在NF基體上。不同倍數下PS-NMO/NF的SEM照片(圖3b、3c)顯示,NMO/NF依次經硫化、磷化處理后,棱角分明的納米棒團簇結構轉變成表面粗糙的類珊瑚球狀結構,表明在NMO表面有非晶相生成。我們在前期工作中發現,磷化作用會在NMO表面生成大量非晶相含磷物種,但不會明顯改變NMO的納米棒形貌[13]。因而,PS-NMO/NF的結構變化應該主要歸功于硫化作用。非晶相類珊瑚球狀多級結構保持了良好的傳質擴散性能,而且提供了大量的活性位點,將有助于提高PS-NMO/NF的催化活性。EDS元素分布圖(圖3d~3h)顯示,PS-NMO/NF表面含有Ni、Mo、O、S和P元素,而且呈現均勻分布。

圖3 NMO/NF(a)和PS-NMO/NF(b、c)的SEM照片;(c)中矩形部分的元素分析圖(d~h)Fig.3 SEM images of NMO/NF(a)and PS-NMO/NF(b,c);Corresponding element mapping images(d-h)in the rectangular area of(c)
圖4為自支撐電極表面S-NMO和PS-NMO粉體的TEM和HRTEM照片。由圖4a可見,棒狀結構的S-NMO表層覆蓋有一層不均勻厚度的薄層。如圖4b所示,該薄層主要由無定形相構成,并在無定形相中分散著結晶度不高的結晶相。根據晶面間距0.24 nm判斷該物種應屬于多硫金屬化合物,但在XRD圖中無明顯衍射峰出現,因此該物相尚不清楚。另外,在S-NMO的HRTEM照片中,可以看到在體相中有晶面間距為0.88 nm的晶格條紋存在,根據標準卡片PDF No.13-0128可知,該晶格結構對應NiMoO4·H2O在10°處的衍射峰,與文獻報道相一致[15]。S-NMO經300℃磷化處理后,得到的PS-NMO仍然保持著棒狀結構(圖4c)。然而P的摻入導致了表層鉬酸鎳晶格形變,以致結構扭曲的晶格條紋無序地散落在無定形相的薄層中(圖4d),該晶面間距為0.65 nm,根據PDF No.12-0348推斷,這歸屬于β-NiMoO4位于14°處的衍射峰。另外,在體相中可以清晰地觀察到晶面間距為0.47 nm的晶格條紋,該條紋屬于β-NiMoO4位于19°處的衍射峰。由以上結果可知,PS-NMO電催化劑體相主要由NiMoO4·H2O和β-NiMoO4構成,經硫化、磷化處理后表面形成以非晶相為主的外延活化層。外延活化層中無定形物相與散落在其中的且結晶度不高的晶相構成了豐富的異質界面。該異質結構與變形的晶格結構為電解水析氫過程提供了豐富的活性位點[16-18]。

圖4 S-NMO(a、b)和PS-NMO(c、d)的TEM和HRTEM照片Fig.4 TEM and HRTEM images of S-NMO(a,b)and PS-NMO(c,d)
采用XPS進一步分析了NMO、S-NMO、PS-NMO粉體的表面組成和化學價態,結果見圖5。如XPS全譜圖所示(圖5a),與NMO相比,S-NMO和PS-NMO表面除了含有Mo、Ni、O元素之外,還分別增加了S和S、P元素,進一步證明S、P元素與NMO表面發生了相互作用,并存在于NMO表面,該結果與EDS相一致。PS-NMO的P2p譜圖(圖5b)顯示,位于133.9和134.8 eV處的特征峰分別歸屬于磷酸鹽中P—O鍵的P2p3/2和P2p1/2軌道[19],而在129 eV左右無峰出現,表明P摻雜到S-NMO中,主要形成了磷酸鹽。S-NMO的S2p譜圖(圖5c)顯示,在168.7處的寬峰屬于S—O鍵[20],表明S進入了NMO晶格形成S—O—M鍵;位于161.7、162.9 eV和163.0、164.2 eV處的2組主峰(峰間距為1.2 eV),分別對應于M—S鍵和M—Sx鍵的S2p3/2和S2p1/2軌道[21-22],表明在硫化過程中S元素與NMO發生了表面反應,生成金屬硫化物和多硫化物。在進一步磷化后,多硫化物主峰消失,且M—S鍵的特征峰向低鍵能方向移動0.2 eV,表明在磷化過程中NaH2PO2·H2O分解產生的H3P與硫化物和多硫化物發生相互作用,將多硫化物中富余的S移除,并使高價金屬還原,導致M—S鍵能紅移。該結果證明了P與硫化物間存在相互作用,并形成了異質結構。O1s譜圖(圖5d)展現出相似的變化規律。如NMO的O1s譜圖中位于530.8 eV處的特征峰為M—O鍵的特征峰[22-23],在532 eV處的特征峰對應H—O鍵[19,23]。在硫化過程中,硫化作用將NMO表面羥基脫除生成硫化物,并形成吸附氧物種,對應S-NMO的O1s譜圖中533.0 eV處特征峰[19]。而且,在531.4 eV處出現了明顯的缺陷氧(Ov)特征峰[23],表明在NMO表面形成了大量具有金屬配位不飽和的硫化物,其具有非晶相的特征。在高溫水熱條件下,硫代乙酰胺發生水解生成H2S。H2S將表面金屬還原引起M—O鍵特征峰向低能態方向遷移0.2 eV。在進一步磷化作用下,該缺陷氧結構傾向于吸附PO43-基團,消耗了表面氧缺陷,生成磷酸鹽[13,24],因此在532.5 eV處出現PO43-基團的P—O鍵特征峰[25]。另外,P摻入NMO晶格使M—O鍵特征峰發生藍移,而S—O鍵及表面吸附氧物種因發生磷化還原和脫硫反應被移除,該現象與S2p譜圖結果相對應。
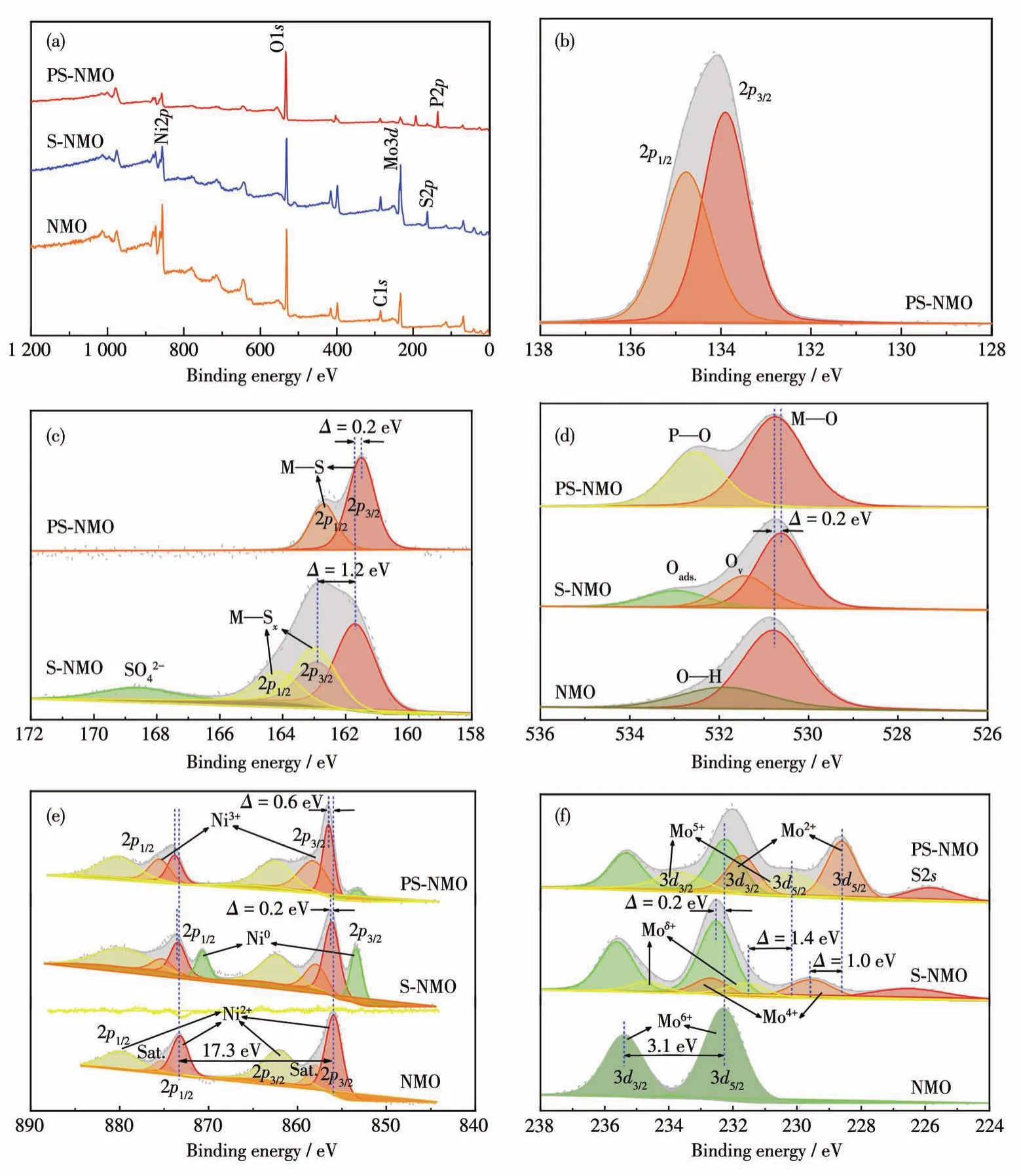
圖5 NMO、S-NMO和PS-NMO粉末的XPS全譜圖(a)、P2p(b)、S2p(c)、O1s(d)、Ni2p(e)和Mo3d(f)譜圖Fig.5 XPS survey(a),P2p(b),S2p(c),O1s(d),Ni2p(e)and Mo3d(f)spectra of NMO,S-NMO,and PS-NMO powders
如上所述,在硫化和磷化過程中主要發生成鹽反應和還原反應,這2個過程在Ni2p和Mo3d譜圖中也得以體現。如圖5e所示,NMO在855.9、862.1 eV和873.2、879.4 eV處出現的2組峰分別屬于Ni2p3/2和Ni2p1/2的主峰及衛星峰,且2個主峰間的能級間距為17.3 eV,證明NMO晶格中Ni主要以Ni2+為主[11,26],而在858和875.3 eV處出現的特征峰證明有Ni3+存在[19,27]。在硫化過程中,由于S進入NMO晶格,導致Ni2+和Ni3+特征峰整體向高鍵能方向遷移0.2 eV。同時,在853.4和870.7 eV處出現明顯的Ni0特征峰[11],證明表面硫化作用產生大量的硫化鎳化合物。但在進一步磷化作用下,Ni0含量顯著下降,而Ni3+含量顯著增加,且Ni2+和Ni3+特征峰進一步向高鍵能方向移動0.4 eV,結合S2p和O1s譜圖,進一步表明PS-NMO中有高價態磷酸鎳物種生成,進而證明硫化物/磷酸鹽異質結構的形成。圖5f為Mo3d譜圖,由圖可見,在NMO中位于232.3和235.4 eV處的峰間距為3.1 eV的2個主峰分別歸屬于Mo6+的3d5/2和3d3/2[11,13,28-30],證明在NMO中只有Mo6+存在。經硫化處理后,在NMO表面部分Mo6+被硫代乙酰胺水解產物H2S還原,導致在231.6、234.7 eV和229.5、232.6 eV處分別出現Moδ+(5<δ<6)和Mo4+。而且,S摻入NMO晶格中,形成S—O—Mo鍵使Mo6+的特征峰發生藍移,此外在226.6 eV處S2s特征峰的出現也證明有硫化物生成[8]。在磷化作用下,H3P還原誘導Mo向低價態轉變,并向低鍵能方向遷移0.2 eV,而形成的Mo5+和Mo2+的含量明顯增加[13,29-30]。Mo元素的價態變化同樣證明了磷化作用對硫化物的影響,并進一步證實硫化物/磷酸鹽異質結構的形成。綜上所述,硫化、磷化作用得到的PS-NMO表面具有豐富的硫化物和磷酸鹽物種,構成了豐富的異質結構界面。
2.2 電化學性能測試
圖6a為NF、NMO/NF、S-NMO/NF和PS-NMO/NF在室溫下1 mol·L-1KOH溶液中的HER性能。從圖中可見,NMO/NF由于具有較差的導電性(盡管樣品表面經過噴金處理,但是NMO/NF的SEM圖像仍存在明顯的放電現象),表現出最差的HER活性[13,31-32]。通過液相硫化改性后形成的二元金屬硫化物具有低電導率,且Ni和Mo元素間協同作用使NiMoSx物種在邊緣Mo位點的H原子吸附自由能接近于0[33],此外硫化后得到的具有粗糙表面的類珊瑚球結構,提供了大量開放的活性位點,從而顯著提高了HER活性。當電流密度為10 mA·cm-2時,HER過電勢從NMO/NF的240 mV降至約100 mV(S-NMO/NF)。進一步經磷化處理后,P元素以磷酸鹽和摻雜形式存在于PS-NMO/NF中。磷酸鹽物種可作為質子載體,加速質子傳遞[33],而且磷原子摻入硫化物中形成的M—P—S鍵,能夠提高活性位點數量,降低電子結合能,促進電子轉移,進而提高HER活性[34]。因此,PS-NMO/NF的HER活性進一步增加,當電流密度為10 mA·cm-2時,HER過電勢約為93 mV,而且在100 mA·cm-2時,僅為180 mV,該HER性能顯著好于相對應的S-NMO/NF和P-NMO/NF電催化劑(電流密度為10 mA·cm-2時,HER過電勢為148 mV[13]),并與商用催化劑性能相近[35]。圖 6b為 NF、NMO/NF、S-NMO/NF和PS-NMO/NF的Tafel曲線圖,可以發現PS-NMO/NF的Tafel斜率為67 mV·dec-1,明顯低于NMO/NF(116 mV·dec-1)、NF(101 mV·dec-1)、S-NMO/NF(79 mV·dec-1)和 P-NMO/NF(88 mV·dec-1[13])。根據HER的Tafel理論值可知,這些電極的HER過程都遵循Volmer-Heyrovsky機制[36-37],而PS-NMO/NF顯示出較快的HER速率。
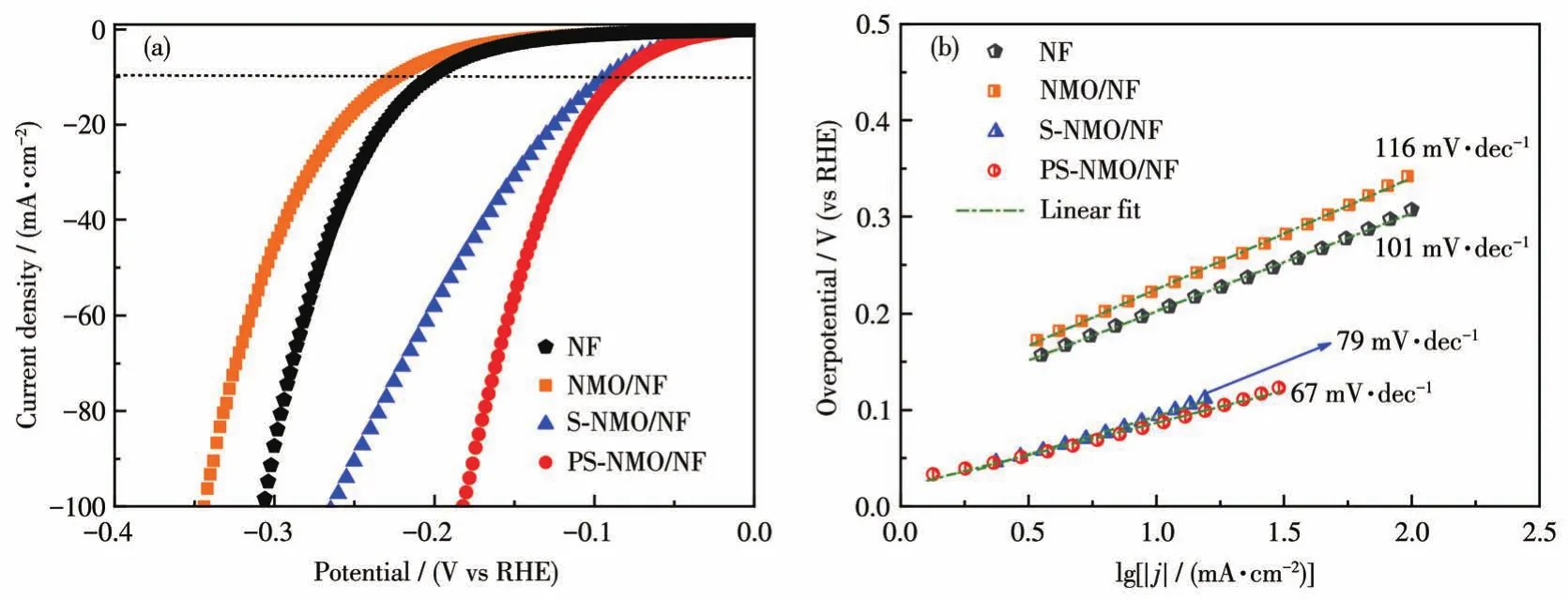
圖6 NF、NMO/NF、S-NMO/NF和PS-NMO/NF在1 mol·L-1KOH中的LSV曲線(a)和Tafel曲線(b)Fig.6 LSV curves(a)and Tafel slopes(b)of NF,NMO/NF,S-NMO/NF,and PS-NMO/NF in 1 mol·L-1KOH
采用CV法測定電極的Cdl值,Cdl值可反映電極的ECSA大小[38-39]。圖7a、7b分別為S-NMO/NF和PS-NMO/NF在非法拉第區域、不同掃描速率下的CV圖,再由CV數據計算出各樣品的Cdl值。由圖7c可知,S-NMO/NF催化劑的Cdl值高達333.88 mF·cm-2,遠遠高于NMO/NF電極的0.67 mF·cm-2。在前期工作中,盡管有大量的非晶相存在,P-NMO/NF的Cdl值僅從NMO/NF電極的0.46 mF·cm-2增加到36.2 mF·cm-2[13]。因此,S-NMO/NF催化劑具有如此高的Cdl值可證明在其表面有大量的非晶相硫化物形成,該結果與SEM和XPS結果相一致。與S-NMO/NF相比,PS-NMO/NF催化劑的Cdl值下降,但仍然高達255.08 mF·cm-2,這是由于在300℃磷化作用下,催化劑表面不穩定含硫物種和易揮發物質被移除以及部分非晶相顆粒發生凝聚。盡管PS-NMO/NF催化劑的Cdl值有所下降,但其HER活性明顯增加,表明磷化作用提高了S-NMO/NF的本征HER性能。
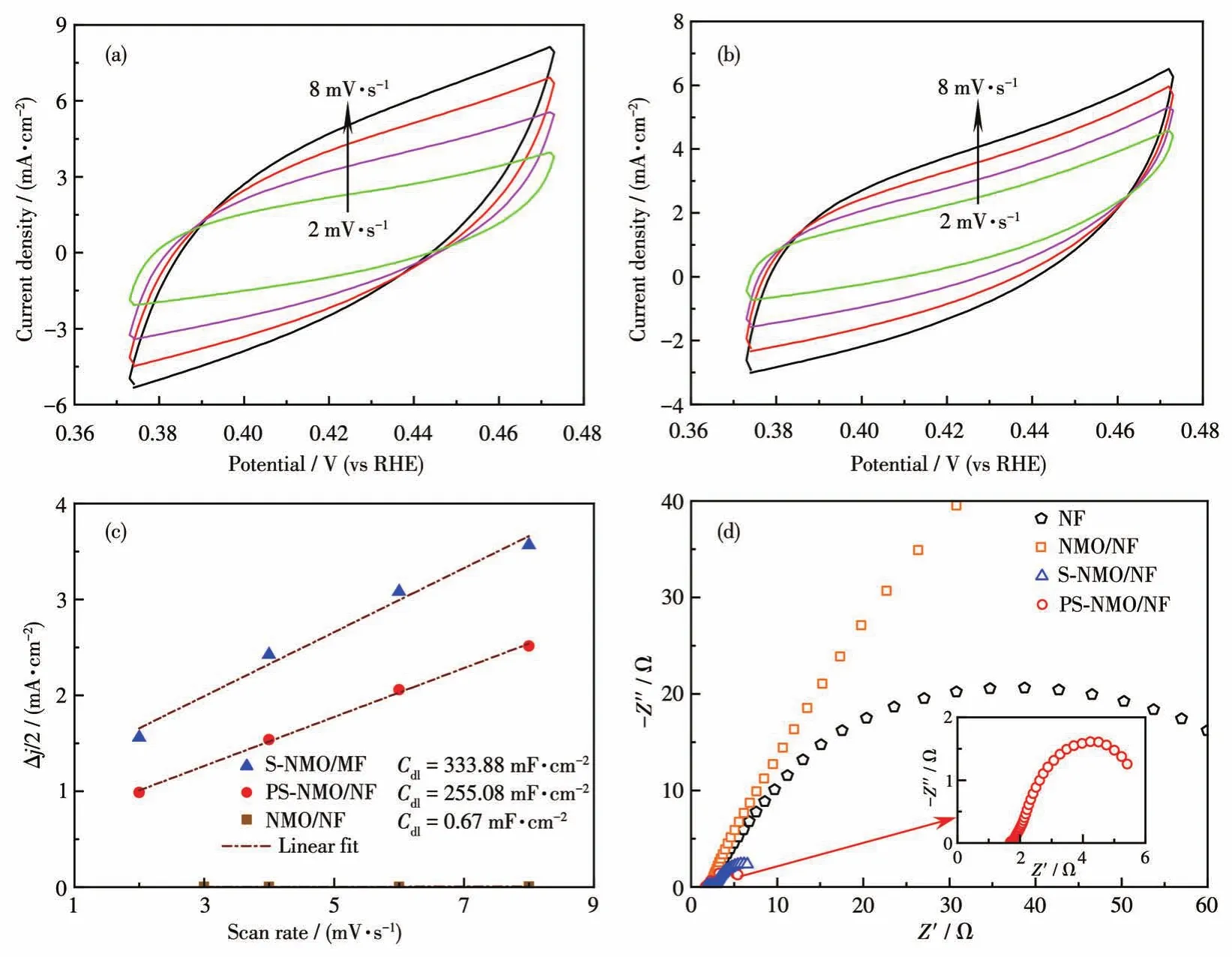
圖7 S-NMO/NF(a)、PS-NMO/NF(b)的CV曲線;NMO/NF、S-NMO/NF和PS-NMO/NF的電流密度隨掃描速率的變化曲線(c);NF、NMO/NF、S-NMO/NF和PS-NMO/NF的EIS譜圖(d)Fig.7 CV curves of S-NMO/NF(a)and PS-NMO/NF(b);Plots of current density as a function of scan rates for NMO/NF,S-NMO/NF,and PS-NMO/NF(c);EIS spectra of NF,NMO/NF,S-NMO/NF,and PS-NMO/NF(d)
在過電勢100 mV下對NF、NMO/NF、S-NMO/NF和PS-NMO/NF進行EIS測試。如圖7d所示,根據Nyquist曲線和EIS擬合數據可知,這些樣品具有相近的溶液電阻(Rs),約為2.0 Ω,而電荷傳遞電阻(Rct)差距明顯。其中,NMO/NF 的Rct值最高(280 Ω),表明了NMO具有較差的導電性,抑制了HER活性[13,31-32]。采用液相硫化處理后,S-NMO/NF的Rct值急劇下降(約7 Ω),證實NMO/NF表面生成的多元硫化物大幅度提高了電荷傳輸效率。再經磷化改性后,獲得的PS-NMO/NF催化劑的Rct值進一步下降至5 Ω,證實了P原子的引入提高了S-NMO/NF導電性,加快了電荷傳遞速率。采用恒電流技術考察了PS-NMO/NF在1 mol·L-1KOH中的HER穩定性。如圖8a所示,在20 h內電流密度沒有明顯衰減,而且穩定性測試后的HER性能顯示當電流密度分別為10和100 mA·cm-2時,過電勢僅增加10和20 mV(圖8b),并且從插圖中可知,反應后的Rct值也沒有明顯增加,表明該催化劑具有良好的穩定性。
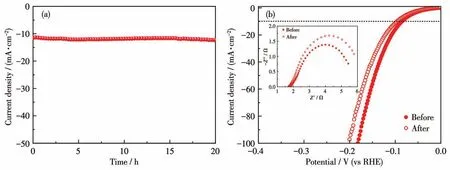
圖8 PS-NMO/NF在1 mol·L-1KOH中的I-t曲線(a)以及HER循環后的LSV曲線和EIS譜圖(插圖)(b)Fig.8 (a)I-t curve,(b)LSV curves,and EIS spectra(Inset)after HER cyclic test of PS-NMO/NF in 1 mol·L-1KOH
3 結 論
先后采用水熱硫化和氣相磷化法處理鉬酸鎳納米棒陣列,獲得具有類珊瑚球結構的自支撐PS-NMO/NF電極。硫化作用使鉬酸鎳納米棒表面生成非晶相硫化物,顯著提高了電化學活性面積,增加了活性位點,并提高了導電性。磷化處理過程使P元素引入硫化物中并形成非晶相磷酸鹽,構建了豐富的硫化物/磷酸鹽異質界面,降低了電荷傳遞阻力,促進了電子轉移,提升了HER性能。在1 mol·L-1KOH電解液中,電流密度分別為10和100 mA·cm-2時,PS-NMO/NF電極的HER過電勢分別為93和180 mV,Tafel斜率為67 mV·dec-1,而且在20 h內可穩定運行。該工作為開發廉價、高效且具有豐富異質界面的電催化劑提供了新思路。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
