注意力特征融合的蛋白質-藥物相互作用預測
2022-09-06 07:30:56李金星馮振華宋曉寧於東軍
計算機研究與發展 2022年9期
華 陽 李金星 馮振華 宋曉寧 孫 俊 於東軍
1(江南大學人工智能與計算機學院 江蘇無錫 214100)
2(薩里大學計算機系 英國吉爾福德 GU2 7XH)
3(南京理工大學計算機科學與工程學院 南京 210094)
(7211905018@stu.jiangnan.edu.cn)
預測蛋白質-藥物相互作用是早期藥物篩選中的關鍵步驟.據美國藥物研究與制造商協會調查,新藥研究成本[1]占用整個制藥業收益的75%.此外,僅有不到5%的經初篩命中的化合物可用于臨床實驗,傳統的篩選方法更是要消耗2~3年的時間,極大程度地耗費了研究人員的精力和時間.借助計算機進行虛擬篩選[2]藥物花費時間短、準確性高,有效降低了該任務的成本.而該方案的核心是依靠計算機預測出蛋白質和藥物之間的相互作用(protein-drug interaction, PDI)進行藥物篩選[3].
預測蛋白質-藥物的相互作用主要包含3個步驟:1)對藥物分子式進行量化并提取其特征;2)對蛋白序列進行量化并提取其特征;3)選擇合適的分類模型,預測藥物和蛋白質是否存在相互作用[4].不難看出,與其他模式識別任務類似,有效提取特征的方法是進行蛋白質-藥物相互作用預測的關鍵.
在提取藥物特征方面,藥物分子量化的理論來源于定量結構與活性關系(quantitative structure activity relationship, QSAR)[5].該關系源自傳統構效關系,并在此基礎上與化學中常見的經驗方程相結合,在藥物化學領域具有廣泛且深遠的影響.該方法把人們對構效關系的認知從定性水平上升到定量水平.從其實際影響來看,定量結構與活性關系揭示了藥物分子與生物大分子結合[6]的模式,指示化合物的某些生物活性可以通過數學模型量化其分子結構特征[7]來獲得,并給出了量化特征的理論依據.在蛋白質特征提取方面,蛋白序列的量化方式[8]主要來源于對氨基酸殘基的特征[9]嵌入.當下主流的做法有自相關矩陣嵌入和殘基序列[10]結合嵌入等.
除了蛋白質和藥物的特征提取方式,分類模型的選擇[11]和設計也是預測蛋白質和藥物相互作用的重要研究內容.現有的預測算法主要分為兩大類:傳統機器學習[12]方法和深度神經網絡[13]方法.傳統機器學習方法包括支撐向量機(support vector machine, SVM)[14]、隨機森林(random forests. RF)[15]、K最近鄰(k-nearest neighbor, KNN)[16]分類算法以及邏輯回歸模型等.基于深度神經網絡的方法主要包括長短期記憶網絡(long short-term memory, LSTM)[17]、卷積神經網絡(convolutional neural networks, CNN)[18]和圖神經網絡(graph neural network, GNN)[19]等.在現有算法中,大多方法都根據靶蛋白種類劃分成4類去解決,即分別在酶、離子通道、G蛋白耦聯受體和核受體蛋白中進行預測.這么做的主要原因是這4類蛋白的類間差異過大,混在一起訓練會使模型變得很難收斂.因此,這些方法訓練出的模型可適用面較窄,往往只局限于預測某一類蛋白和藥物的相互作用.當無法判斷一種新的未知蛋白的真實屬性時,該類方法的魯棒性和實用價值會大大降低.
為解決上述模型泛化能力不足的問題,由Lee等人[20]提出的DeepConv-DTI模型對變長的蛋白序列進行補零定長,將差異較大的蛋白序列特征固定在相同維度空間中,并采用卷積神經網絡提取蛋白質的低維實值特征,有效拓寬了模型的泛化性.與之相似的是,?ztürk等人[21]提出的DeepDTA模型還額外添加配體最大公共結構和蛋白質結構域特征來提升模型的效能.然而,這2個模型提取藥物特征的方法仍然存有不足,僅使用擴展連通性指紋Morgan[22]作為量化藥物的方法無法提取分子的結構信息.Morgan指紋編譯是一種圓形拓撲指紋,先枚舉藥物分子中所有可能的原子位次,然后將各原子周邊的原子信息增加到該原子上形成數組,并散列至一個數字,最后通過計算連通性選擇最合適的位次得到對應的分子指紋.這種方法可有效提供分子特征的細節,但藥物分子結構信息的缺乏導致蛋白質-藥物相互作用預測的性能明顯不足.而CPI-GNN[23]和GraphDTA[24]等模型借助圖卷積網絡提取藥物特征一定程度上獲取了藥物分子的結構特征,在其實驗中取得相對不錯的效果.但是圖卷積模型對分子結構的辨識度會隨著藥物種類的增多而下降,也會致使模型的整體性能下降.
此外,文獻[20-21,23-24]所提的方法大都使用多層感知器(MLP)模型[25]預測蛋白質-藥物的相互作用.該方法無法凸顯藥物特征中重要的局部信息,致使整個模型的預測性能達不到最佳效果.為解決這一問題,Wang等人[26]在2020年初提出了一種使用深度長短期記憶網絡(deep long short-term memory, DeepLSTM)的方法預測蛋白質-藥物相互作用,在酶和G蛋白耦聯受體的作用預測中都取得了最佳效果.雖然這一方法無法有效預測大規模數據下的PDI,但論述了通過添加時序信息可有效捕捉更多的鑒別PDI的特征.2020年Chen等人[27]提出基于自然語言處理(natural language processing, NLP)中Transformer骨干網絡[28]中的TransformerCPI模型.該模型利用自注意力機制有效地捕捉化合物和蛋白質之間的關聯特性,在各公開數據集中均表現良好.但因為其提取的蛋白特征不夠充分導致其在Kinase數據集上的收斂效果不夠理想.
為進一步提升在大規模數據下PDI實驗的預測效果,本文結合前人的工作提出了一種將深度卷積神經網絡與自注意力循環網絡相結合的模型.在藥物特征提取方面,由于Morgan無法提取藥物分子的子結構特征,本文在其基礎上增加了Mol2Vec向量[29]嵌入.Mol2Vec是一種無監督的機器學習方法,用于學習分子亞結構[30]的向量表示.與Word2Vec模型[31]一致,密切相關單詞的向量在向量空間中非常接近,Mol2Vec可以學習指向與化學相關子結構方向相似的分子子結構的向量表示,并以對各個子結構做向量求和的方式來編碼化合物.Mol2Vec模型克服了常見復合特征表示的缺點,如稀疏性和位沖突,在多個化合物特性和生物活性數據集上驗證了其預測能力.為了補充更多藥物特征信息,本文在原基礎上額外添加由消息傳遞神經網絡(message passing neural networks, MPNNs)[32]提供的圖特征,經實驗驗證在數據規模較小的實驗中添加該特征可有效提升模型的性能,但對數據規模較大的實驗模型效果不明顯.據此,本文先用循環網絡采集Morgan指紋和Mol2Vec向量的重要信息并將兩者進行融合,再根據數據規模差異考慮是否融合圖特征,最后本文用密集型卷積網絡進一步提取出藥物分子的特征.
在蛋白特征提取方面,本文根據生物活性對蛋白質的氨基酸序列進行歸類[33],這樣可有效減少蛋白嵌入特征的稀疏性.并借鑒了Lee等人[20]的經驗,對變長的蛋白序列編碼進行補零定長并嵌入至等長的特征空間中.為了模型收斂得更好以及蛋白質特征采集得更詳細,本文選擇密集型卷積網絡DenseNet[34]提取蛋白特征,該模型可直接連接來自不同層的蛋白特征,通過特征重用可有效提升效率和精度.接著利用藥物特征對蛋白特征進行注意力針對訓練,這樣得到的蛋白特征除了包含蛋白序列本身信息之外還包含蛋白藥物的關系信息.其結果與藥物特征一同放入雙方向門控循環單元提取PDI的特征信息.
門控循環單元[35]是循環神經網絡(recurrent neural network, RNN)中的一種門控機制.與其他門控機制相似,門控循環單元旨在解決標準循環神經網絡中的梯度消失(爆炸)問題,并同時保留序列的長期信息.由此得到的PDI特征信息再經過自注意力機制對重要區域進行加權,以及經過全連接層映射成長度為2的1維向量.最后放入Softmax激活函數中進行歸一化處理作為蛋白質-藥物相互作用的預測結果.除此之外,本文結合所提模型設計了可用于藥學研究的界面,并提供了篩選抑制乙酰膽堿脂酶(AChE)和丁酰膽堿酯酶(BuChE)藥物的方法.這類藥物對治療阿爾茲海默癥具有重要意義[36],本文也借此進一步闡述了模型的實用價值.
綜上所述,本文的主要創新點包括:
1) 提出一種復合提取藥物特征的方法,將Morgan指紋編譯、Mol2Vec向量以及MPNNs圖結構特征合理融合,豐富了藥物特征信息;
2) 根據生物活性對氨基酸進行歸類,有效降低蛋白特征的稀疏性,采用密集型卷積網絡提取特征,實現特征重用,提升模型的效率和精度;
3) 發現深度網絡結合門控循環單元提取蛋白質和藥物特征,結合能強化蛋白質和藥物關系特征的注意力網絡,可有效提升模型的性能.
1 本文方法
1.1 藥物特征表示
藥物特征提取是預測PDI的重要環節.其目的是提取藥物的鑒別特征,使得分類器可以更好地理解藥物性質并能區分出不同藥物之間的差異.因此,有效的藥物特征需要具備可鑒別性和典型性,且類間的距離相對較遠而類內的距離相對較近.這樣便于提升分類模型的預測精度.常用的藥物特征嵌入方法有2種,包括Morgan指紋和Mol2Vec向量.
Morgan指紋又稱擴展連通性指紋,是一種圓形指紋.借助Morgan指紋編碼藥物分子式如圖1所示:
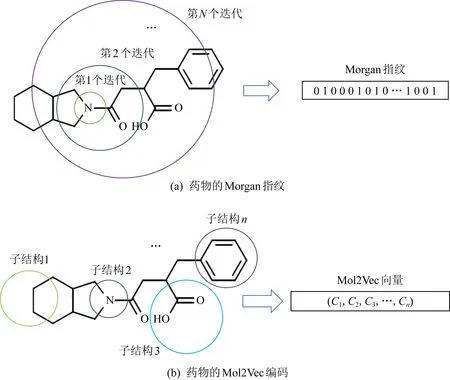
Fig. 1 Drug feature embedding圖1 藥物特征嵌入
藥物特征嵌入的步驟為:首先根據給定的半徑分析每個原子的環境和連通性;然后散列編碼所有可能形成的結構;最后根據散列算法將編碼信息放縮到預定長度.這種方法的代表性雖然很全面,但由于尺寸較大且信息過于離散,難以合理地表達藥物的子結構信息.另一種常用的嵌入藥物分子式特征的方法是Mol2Vec向量編碼.Mol2Vec是從NLP中的Word2Vec演變而來,可以學習分子間化學性質指向相似的子結構信息.其編碼方式為將各個子結構向量的和作為化合物的特征向量.該方式可將藥物的子結構特征表示得很清晰,具有很強的典型性,也是對Morgan特征的一項重要補充.
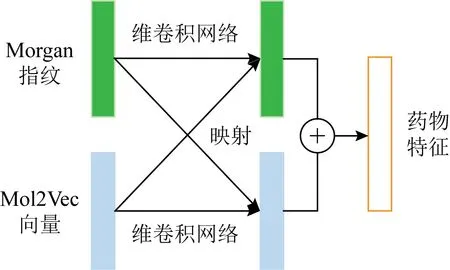
Fig. 2 The feature fusion module圖2 特征融合模塊
為了提取具備鑒別性和典型性的特征,本文將圖1所示的2種藥物特征嵌入方法結合使用.首先,通過雙向門控循環單元對嵌入特征進行特征抽取;然后將2種方法所得藥物特征按照圖2的方式融合;最后將融合特征放入密集型卷積網絡特征抽取,作為與蛋白結合預測的藥物特征.其中,融合方法為先用1維卷積(1×3)將原特征轉至下層特征,隨后使用映射矩陣分別將Morgan特征和Mol2Vec特征映射到與對方相同的特征空間中并與下層特征相加,2個映射矩陣互為轉置,公式為:
FM1=Conv1d(FM0)+VFN0,
(1)
FN1=Conv1d(FN0)+VTFM0,
(2)
其中,FM0和FM1分別是Morgan原特征與下層特征,FN0和FN1分別是Mol2Vec原特征與下層特征,V是可訓練的映射矩陣,其維度分別是FM0和FN0的長度.使用同映射矩陣可以使2類特征的關聯性更強、模型更容易收斂,最后將兩者下層特征相加得到融合特征.
同時,本文發現消息傳遞網絡可以抽取藥物的平面結構特征,存有提升實驗效果的可能.MPNNs由Google的研究人員提出并用于預測量子化學性質,可有效應用于小樣本模型[37].具體算法為:首先,構造初始狀態集,每個狀態用于圖中的每個節點;然后,使每個節點與其鄰居交換信息以進行消息傳遞,這樣每個節點狀態將包含對其直接鄰居的感知.重復這2個步驟,每個節點便可獲得其2階鄰域的信息,依此類推.達到預想次數的“消息回合”,便可將所有上下文的節點的狀態轉換表征為整個圖的特征.其節點更新權重的公式為
(3)
(4)
如圖3所示,本文嘗試采用特征融合模塊將MPNNs模塊所提取的藥物圖特征與由Morgan和Mol2Vec融合的特征相融,然而實驗效果在不同規模數據集上的表現有所差異,具體的內容將3.2節進行詳細的論述.實驗表明,MPNNs提供的圖結構特征會讓模型在小規模數據集上表現得更好,但對數據規模較大的實驗效果不明顯.
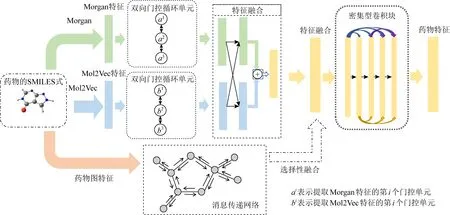
Fig. 3 The drug feature extraction model圖3 藥物特征提取模型
1.2 蛋白質特征表示
本文沿用Lee等人[20]的方法對蛋白質一級結構氨基酸殘基序列進行編碼,將字符形的序列嵌入到離散的整形向量中.本文對蛋白質序列做了一步預處理,在本文所提各數據集中蛋白質序列是由22類氨基酸混合組成,若以此作為文本,3個氨基酸片段作為詞劃分蛋白,便有22×22×22=10 648種組合,這便導致蛋白的特征矩陣過于稀疏.為解決這一問題,本文借用Che等人[33]使用的方案,根據生物化學的特性將22種氨基酸歸類為6種:a={H,R,K},b={D,E,N,Q},c={C,X},d={S,T,P,A,G,U},e={M,I,L,V},f={F,Y,W}.
這樣序列MSPLNQSAEGLPQEASNRSLN便可以轉化為eddebbddbdedbbddbadeb,該方法得到組合數為6×6×6=216種可顯著降低特征矩陣的維數.同時,本文使用DenseNet提取蛋白特征.DenseNet提出了一個非常激進的密集連接機制,即互相連接所有的層.具體來說就是每個層都會接受其前面所有層作為其額外的輸入,每層都會與前面所有層在維度上連接在一起并作為下一層的輸入.對于一個L層的網絡,DenseNet共包含(L+1)L/2個連接.如圖4所示,直接連接來自不同層的蛋白特征可有效提升實驗的效率和精度.
本文采用1維卷積從上層模型提取特征,卷積提取特征的公式為

(5)
其中,函數x(t)和q(t)表示卷積的變量,p表示積分變量,t表示使函數q(t-p)位移的量,*表示卷積.蛋白質序列信息在經過特征嵌入、密集型卷積網絡、最大池化、全連接后轉變為128維的特征向量,與藥物的特征向量一同放在分類模型中以預測PDI結果.
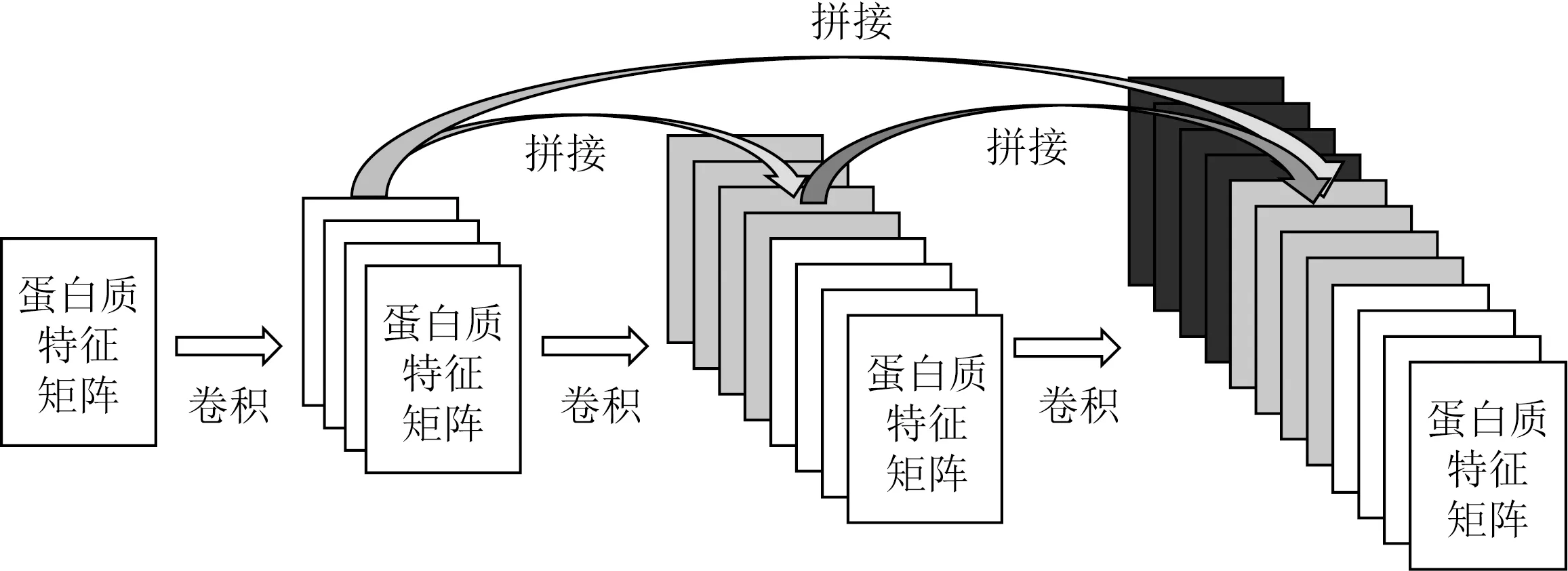
Fig. 4 The dense protein feature extraction model圖4 密集型蛋白特征提取模型
1.3 模型整體框架
本文方法的整體框架如圖5所示.該框架由3個部分組成:蛋白質、藥物特征提取模塊以及預測PDI模塊.本文采用的是端到端的訓練模型,即分類器與2個特征模型的訓練任務同時進行.流程包括:
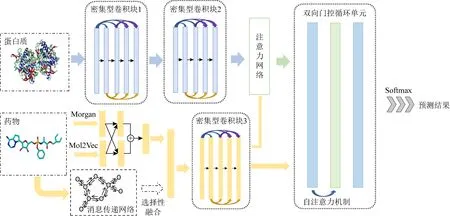
Fig. 5 The overall framework of the proposed method圖5 本文方法框架
1) 藥物分子經過2種特征嵌入后,由雙向門控循環單元和鄰域卷積先后抽取藥物特征,并對由卷積模塊所提取的蛋白特征做注意力權值增強.具體做法即給定一個藥物分子特征向量Fdrug和蛋白質子序列特征向量P=(P1,P2,…,Pi),隨后為其構造一個關于Fdrug的注意力矩陣.根據不同注意力權重來計算不同子序列對藥物分子的重要性.公式為
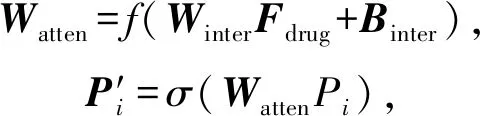
(6)

2) 拼接蛋白質特征和藥物特征,并用自注意力機制[38]對PDI信息進行加權提取,具體做法即給定拼接后PDI特征向量cinter,訓練一個自注意力矩陣Wself-atten對相互作用信息區域進行加權學習,公式為
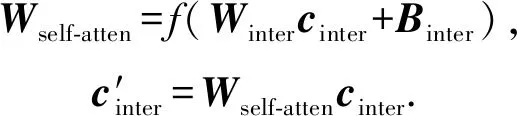
(7)
本文將在3.2節對2塊注意力機制的使用效果進行驗證.
1.4 損失函數和模型優化
本文使用交叉熵損失作為訓練模型的損失函數:
(8)
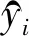
為了避免發生過擬合的情況,本文用L2范式作為懲罰項來約束模型的優化:
(9)
其中,W和b是模型全局的權重和偏置,λ是懲罰因子.同時本文嵌入dropout層[40]來輔助解決這個問題.為兼顧訓練的效率和分類的精度,本文用Adam優化器[41]來更新模型的權值.
2 數據集
本文使用BindingDB,Kinase,Human,C.elegans這4個數據集來驗證模型的效果.BindingDB是從大量科學文獻中采集的可公開訪問的蛋白質藥物相互作用數據庫.在2018年,由Gao等人[42]按照以下規則從該數據庫中采集39 747個正樣本和31 218個負樣本制成用于評估PDI模型的BindingDB數據集.規則為:
1) 所記錄的藥物分子具有化學標識符(PubChem CID)以及以smiles表示的化學結構;
2) 所記錄的蛋白質也需具有數據標識符(Uniprot ID)以及序列表示和基因本體注釋;
3) 記錄具有IC50值,即相互作用的主要指標;
4) 因為專注于小分子藥物,所有的化學分子量均需小于1 000道爾頓;
5) 遵循Wang等人關于活動閾值的討論,若IC50小于100 nm則記為正,IC50大于10 000 nm則記為負.
該數據集按如表1的方案來劃分訓練、驗證和測試的子集,其中驗證和測試的子集包含訓練集中未被觀察到配體或蛋白質的PDI樣本.因此,BindingDB數據集可以評估模型對未知藥物和蛋白質的魯棒性.
Kinase數據集是由Chen等人[27]基于KIBA數據集構建而成,包含229個蛋白樣本和1 644個藥物樣本.KIBA數據集已涉及多種用于測試活性的評價機制.對比多種生物活性評分,可有效減少由人為因素給數據集造成的偏差.同時Kinase的負樣本遠多于正樣本,如表2所示.
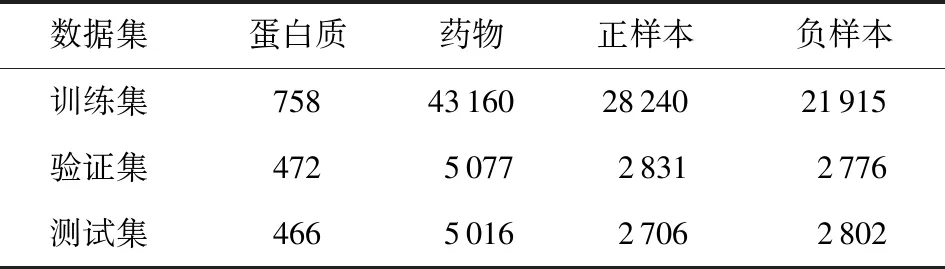
Table 1 The Distribution of BindingDB Dataset表1 BindingDB數據集分布

Table 2 The Distribution of Kinase Dataset表2 Kinase數據集分布
用此數據集可有效測試模型在樣本不平衡下的性能,而在實際藥物篩選過程中,沒有相互作用的樣本顯然比有相互作用的樣本多得多,因此模型在不平衡數據集下的表現也十分重要.
數據集Human和C.elegans是由Tsubaki等人[23]在2019年根據前人的工作匯總而成,其中Human數據集涵蓋852種人類蛋白以及1 052種藥物分子,數據共存有3 369個正樣本和2 843個高度可信的負樣本.C.elegans數據集包含2 504種線蟲蛋白和1 434種藥物分子,數據共存有4 000個正樣本和3 511個高度可信的負樣本.然而這2個數據集沒有劃分出訓練和測試的子集,因此本文采用交叉驗證的方式來評估模型在這2個數據集上的表現.本文所有數據及所提出的模型程序[43]均可從github上獲取.
3 實驗結果與討論
3.1 實驗環境以及參數設置
本文實驗所配置的硬件:采用的中央處理器和顯卡分別為Intel Core i7-8700k和NIVADA GeForce RTX 2060 s,并配用Windows10的操作系統.本文在Python3環境下的Keras深度學習框架上訓練和評估模型,并使用Sklearn等機器學習工具處理實驗數據.訓練模型時,超參數的設置對模型優化有很大影響.本文優先固定參數優化的學習率,在此基礎上使用網格搜索法對其他各參數進行尋優.多輪實驗最終確定了模型的超參數設置,如表3所示:
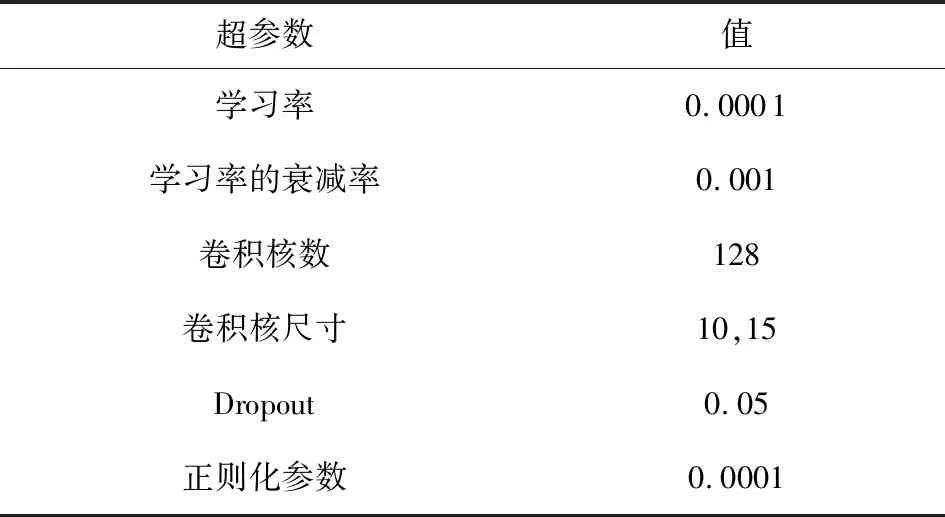
Table 3 The Hyper-Parameter Settings表3 超參數設置
本文使用2個指標評估各模型:
1) ROC曲線下的面積AUC.ROC曲線上的每個點是由2個指標的值來確定坐標,即真陽率(true positive rate,TPR)和假陽率(false positive rate,FPR),TPR=TP/(TP+FN),FPR=FP/(TN+FP).其中TP表示陽例預測為正例子的個數,FP表示陰例預測為正例子的個數,TN表示陰例預測為負例子的個數,FN表示陽例預測為負例子的個數.
2) PR曲線下的面積AUPR.PR曲線上的每個點是由2個指標的值來確定坐標,即精準率(precision,P)和召回率(recall,R),P=TP/(TP+FP),R=TP/(TP+FN).
3.2 消融實驗
本文仿照DeepconvDTI的方法提取藥物特征,即藥物的表征由Morgan向量構成.結合本文所提模型在BindingDB數據集上的訓練,在測試集上的最優結果的AUC=0.954.經調研發現,僅用Morgan向量表征藥物易于忽視分子的亞結構信息,而這部分內容對量化藥物特征信息十分重要.因此,本文使用涵蓋藥物亞結構信息的Mol2Vec向量對Morgan向量進行補充,并用DenseNet多尺度提取分子特征進一步優化了模型,最優結果AUC=0.963.該結果證實了藥物特征提取方法的優化對模型整體預測能力的提升具有積極意義.
Riba等人[37]在研究圖網絡主動學習分子隱式結構特征的過程中表明:消息傳遞神經網絡(message passing neural networks,MPNNs)可以幫助模型學習分子的結構信息.在此基礎上,本文試圖添加分子平面結構圖的特征來進一步優化模型,并在原框架基礎上添加MPNNs模塊,然而在BindingDB數據集上測試效果相比原先模型下降了0.5%,這并不符合本文的預期.經仔細查閱發現,在Riba等人的工作中,數據的規模相對較小,于是本文在Human和C.elegans數據集上做了相同的消融實驗,結果如表4所示:
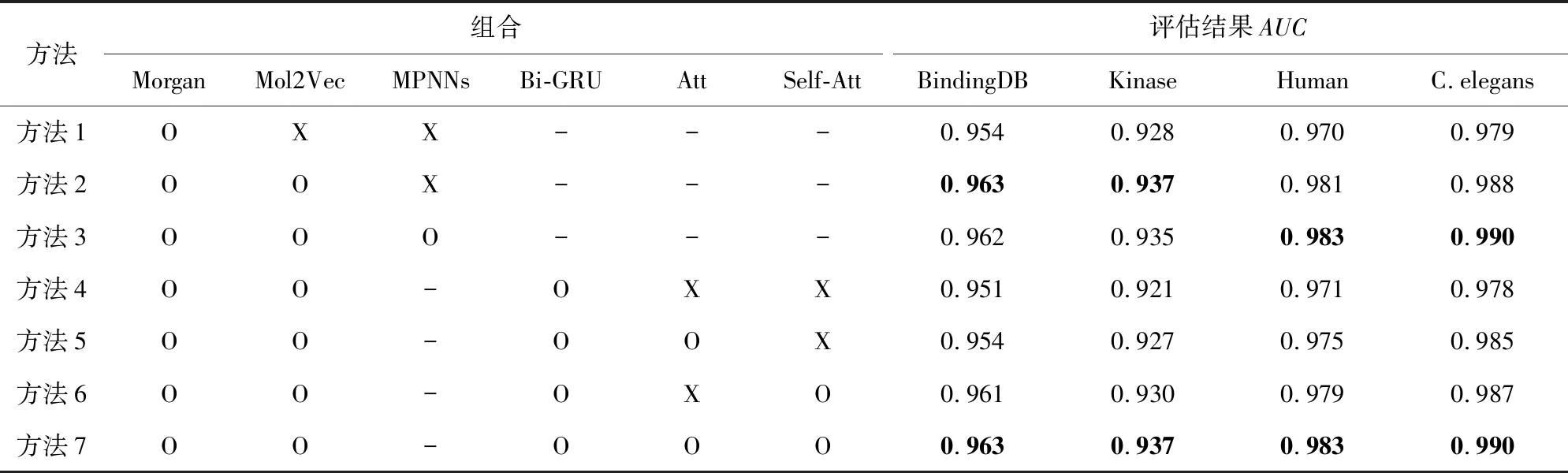
Table 4 The Ablation Study Based on Four Datasets表4 基于4個數據集上的消融實驗
實驗發現,添加MPNNs模型,會使模型整體在Human數據集和C.elegans數據集上提升0.2%,而在規模相對較大的BindingDB數據集和Kinase數據集上的效果分別下降了0.1%和0.2%.對此本文根據訓練周期(epoch)的更迭,在BindingDB和Human數據集上繪制了不同藥物特征組合下的樣本類內距離和類間距離的變化趨勢圖.樣本類內距離和類間距離計算公式為
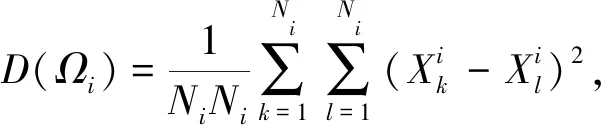
(10)
(11)
根據圖6所示,在數據規模相對較大的BindingDB數據集中,僅使用Morgan指紋和Mol2Vec向量結合量化特征,會讓樣本隨著epoch的迭代類間距變得更遠、類內距離變得更近,這為分類器的判別提供了很大的便利.而添加MPNNs模型后在BindingDB數據集上的效果略微下降,但在Human數據集上的效果卻有所提升,也從側面論證了MPNNs的介入有益于模型在中小規模數據下的收斂,但對數據規模較大的實驗效果不明顯.
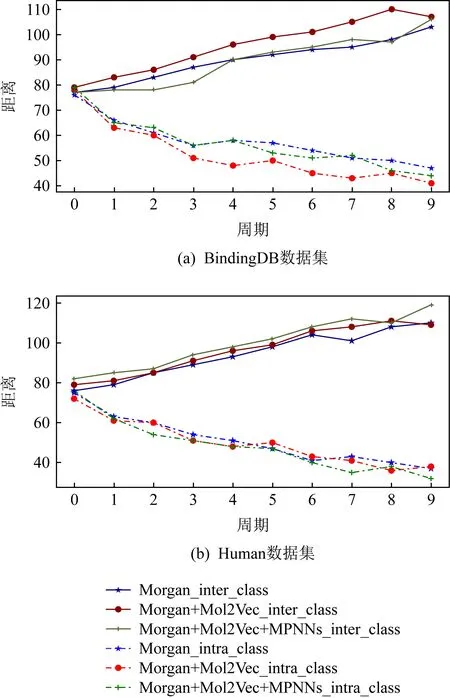
Fig. 6 The trend of intra-class distances and inter-class distances圖6 類內距離和類間距離趨勢圖
確定了提取藥物特征的模塊后,據多輪實驗結果發現,使用雙方向門控循環單元做分類模塊的性能最佳.在此基礎上,本文開始研究如何利用注意力模塊增強模型對重要信息的感知.如表4的方法4~7所示,模型在BindingDB測試集上的初始AUC=0.951.本文試圖增強蛋白特征與對應藥物特征的關聯性,便在兩者之間添加注意力模塊,其結果相比初始AUC提升了0.3%.在此基礎上,本文通過自注意力模塊對合并的特征向量進行重要信息加權將模型的性能提升到最佳,其測試的AUC=0.963.除此之外,本文也附加僅添加自注意力模塊的實驗和在其他數據集上的測試結果,發現在各數據集上自注意力模塊都可以使模型的精度提升近1%.
3.3 對比實驗
本文用4個數據集檢驗所提模型,并與前人所提的同類模型進行對比實驗,其中包括Tsubaki工作中提到的最近鄰模型(KNN)、隨機森林(RF)、L2邏輯回歸、支持向量機以及CPI-GNN模型,不過因為在文獻[23]中未涉及除了CPI-GNN以外的各個模型的參數細節,所以除了Human和C.elegans數據集外本文不再討論前4類模型的性能.此外,本文還添加由Nguyen等人[24]提出的GraphDTA模型、由Lee等人[20]提出的DeepConvDTI模型以及由Chen等人[27]提出的GCN模型和TransformerCPI模型的討論.值得注意的是,這些模型均是近年來PDI預測的典型模型.
如表5所示,本文依次在BindingDB數據集上比較提到的GraphDTA,GCN,CPI-GNN,Trans-formerCPI,DeepConvDTI和本文所提模型,前4個模型結果均從文獻[27]中獲取,DeepConvDTI模型與本文所提模型的結果都是在實驗中經過調整參數所得到的最優結果.從表5可清晰地發現,本文所提模型的預測結果比當前前沿模型更佳;AUC較基線水準提升了0.019,與TransformerCPI相比提升了0.012.
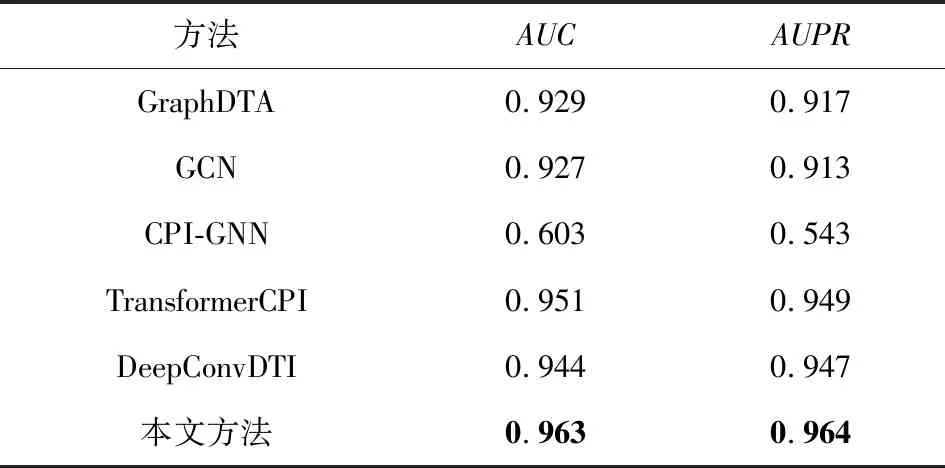
Table 5 Comparative Experiments on BindingDB Dataset表5 BindingDB數據集上的對比實驗
在實際應用中,蛋白質和藥物相互作用的負例個數遠超正例個數,因此控制負例樣本遠多于正例樣本更貼合實際情形.為驗證模型在這一情形上的性能,本文在Kinase數據集上與文獻[27]所涉及的各個模型進行對比.在第2節詳細介紹了Kinase數據集因樣本不平衡問題導致很多模型在此數據集上很難收斂.而從表6可以發現,與另外4個模型相比,本文方法在仿真的不平衡數據集上的表現依舊優異,具備突出的實際應用價值.
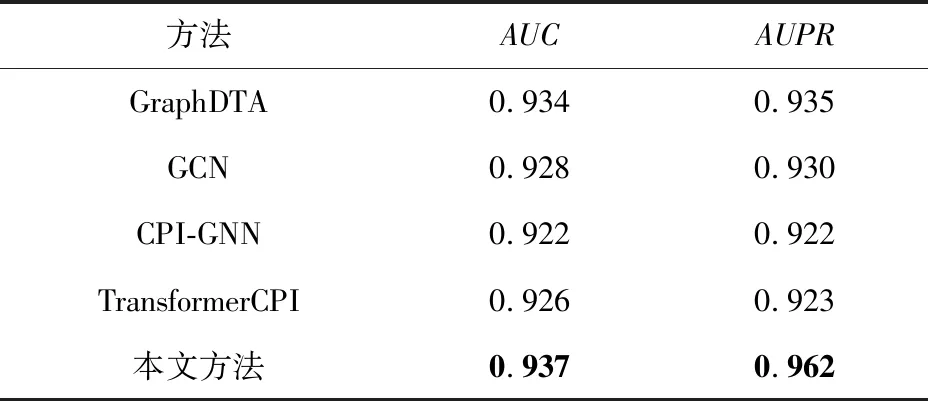
Table 6 Comparative Experiments on Kinase Dataset表6 Kinase數據集上的對比實驗
最后,本文再次在廣泛使用的Human和C.elegans數據集上評估新模型的性能.由于這2個數據集未劃分出用于訓練和測試的子集,本文采用交叉驗證的方式評估各個模型.為了證明實驗結果具備比較價值,本文劃分數據的方式與前人相同,即按4∶1的比例劃分訓練集和測試集,評估體系也與Tsubaki等人[23]保持一致.本文將數據次序打亂隨機劃分了10次,實驗結果的均值和方差分別如表7和表8所示.結果足以證明,本文方法在精確度以及穩定性上相比其他同類模型都更為優異.
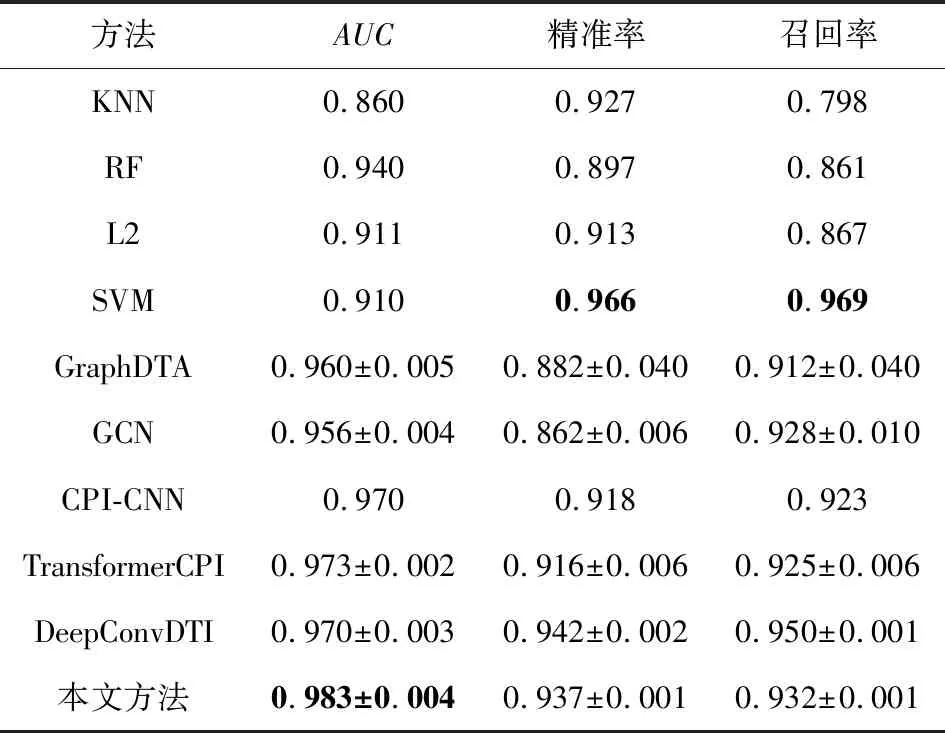
Table 7 Comparative Experiments on Human Dataset表7 Human數據集上的對比實驗
因為C.elegans數據集樣本種類豐富、樣本彼此的相似度很高,所以各深度模型在該數據集上的表現都很好.但從表8可以看出,在交叉驗證下,本文所提模型在該數據集上的表現更為穩定一些.
與當前前沿模型相比,本文所提模型在4個公開數據集上均能取得最佳的識別效果,一定程度上可以驗證其優越性.為了更好地展示模型的實際價值,本文將在3.4節根據具體的醫學案例,介紹所提模型的使用方法并給出仿真實驗的結果.
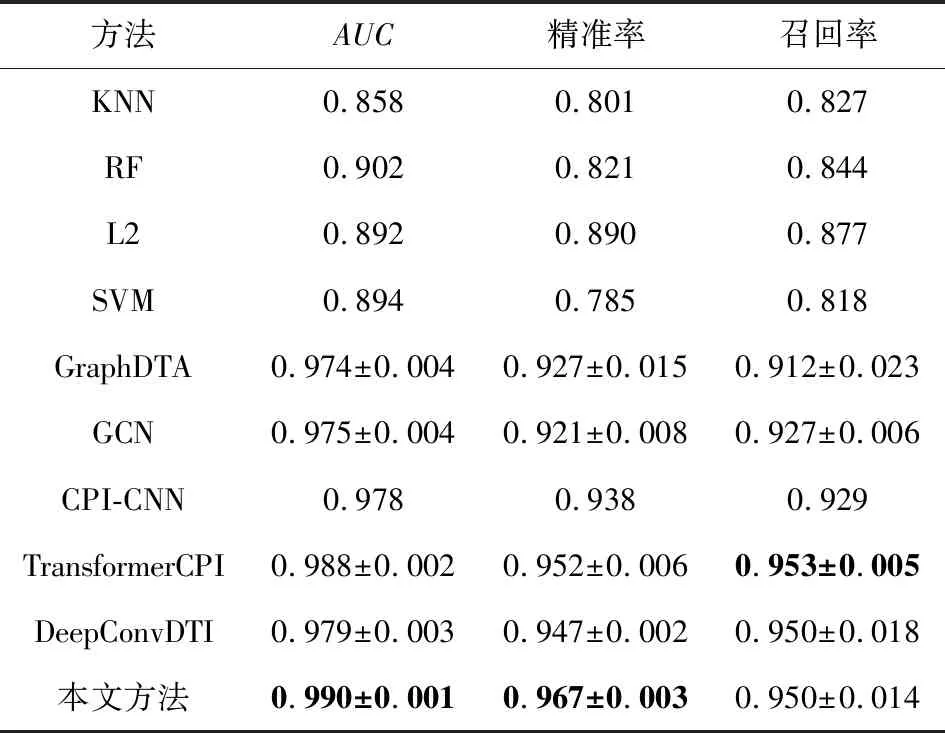
Table 8 Comparative Experiments on C.elegans Dataset表8 C.elegans數據集上的對比實驗
3.4 模型在篩選治愈阿爾茲海默癥藥物的應用
癡呆癥是公共衛生管理中最值得關注的問題之一,而80%以上的癡呆癥患者患有阿爾茨海默病(AD)[44].然而,當前普及的治療方案僅可起緩解作用,無法根據病理學使患者痊愈或逆轉患者的疾病進程.因此,當前醫學界仍急需一種可延緩或阻止疾病進展的新型治療方案.神經遞質乙酰膽堿(ACh)的減少導致膽堿能神經元的丟失在醫學界是公認的AD病因,而通過抑制乙酰膽堿酯酶(AChE)可以提升ACh的水平,以此提高患者的認知能力[43].
雖然乙酰膽堿酯酶(AChE)在疾病晚期的活性會降低,但與圖7所示的AChE結構類似的丁酰膽堿酯酶(BuChE)含量在疾病晚期會有增加的可能[36],而BuChE的含量增加也會水解ACh從而再次加重患者的病情.敲除乙酰膽堿酯酶基因的小鼠實驗[45]支持了這一假設,也證實了通過抑制BuChE可以改善認知能力以及提升患者的記憶力.因此,尋找抑制AChE和BuChE的靶向藥物是治療AD的關鍵方向.根據這一理論,本文將所提出的模型設計成一套可應用的藥物篩選工具[46],并用之篩選抑制AChE和BuChE的藥物.具體操作步驟:1)將待測蛋白質序列數據與大量的藥物分子式數據同時放入篩選工具中;2)系統會根據訓練好的模型對藥物進行篩選;3)系統會給出Top15的藥物序號,并將這些藥物與待測蛋白質相互作用的預測結果以直方圖的形式自高而低展現.為了證實文本所提模型的價值,在該項任務中選擇用于測試的藥物分子均不在訓練集中,但需要指明的是,訓練集中包含AChE的序列數據但不包含BuChE的序列數據,而AChE序列與BuChE序列相似性為65%.當然,PDI預測模型的核心原理是通過學習已有的蛋白質藥物相互作用關系并從中擬合出一個規律函數來推斷未知的相互作用關系.所以,如果測試的樣本與訓練的內容毫無關聯,那么其推斷的依據便失去了理論支撐.
Fig. 7 Mimic diagram of protein-drug interaction圖7 蛋白質-藥物相互作用模擬圖
本文借用Kumar等人[47]提供的藥物數據作為測試的目標,測試數據中包含的35種化合物是由Kumar等人[47]在Asinex庫中手動篩選出的,具有高度可信性.文獻[47]中給出了藥物分子的2維結構式以及Asinex編號,本文通過相關編號以及藥物的結構式在PubChem上檢索出用于預測PDI的SMILES式.同時,Kumar給出了這部分藥物分子對AChE和BuChE的抑制率(inhibition rate,IR),本文按照其定義的準則,將IR<0.5記為無相互作用,IR>0.5記為有相互作用,獲得的最終測試數據如表9所示.
值得注意的是,測試藥物皆不存在于訓練集中,但訓練集中不排除具有相似結構以及相同官能團的藥物分子.根據系統的預測結果,本文按預測值高低將分子序號排列,得到Top15的直方圖.如圖8所示,其中灰色直方圖表示在原數據中該藥物與測試酶具有相互作用,條紋直方圖表示原數據中該藥物與測試酶沒有相互作用.
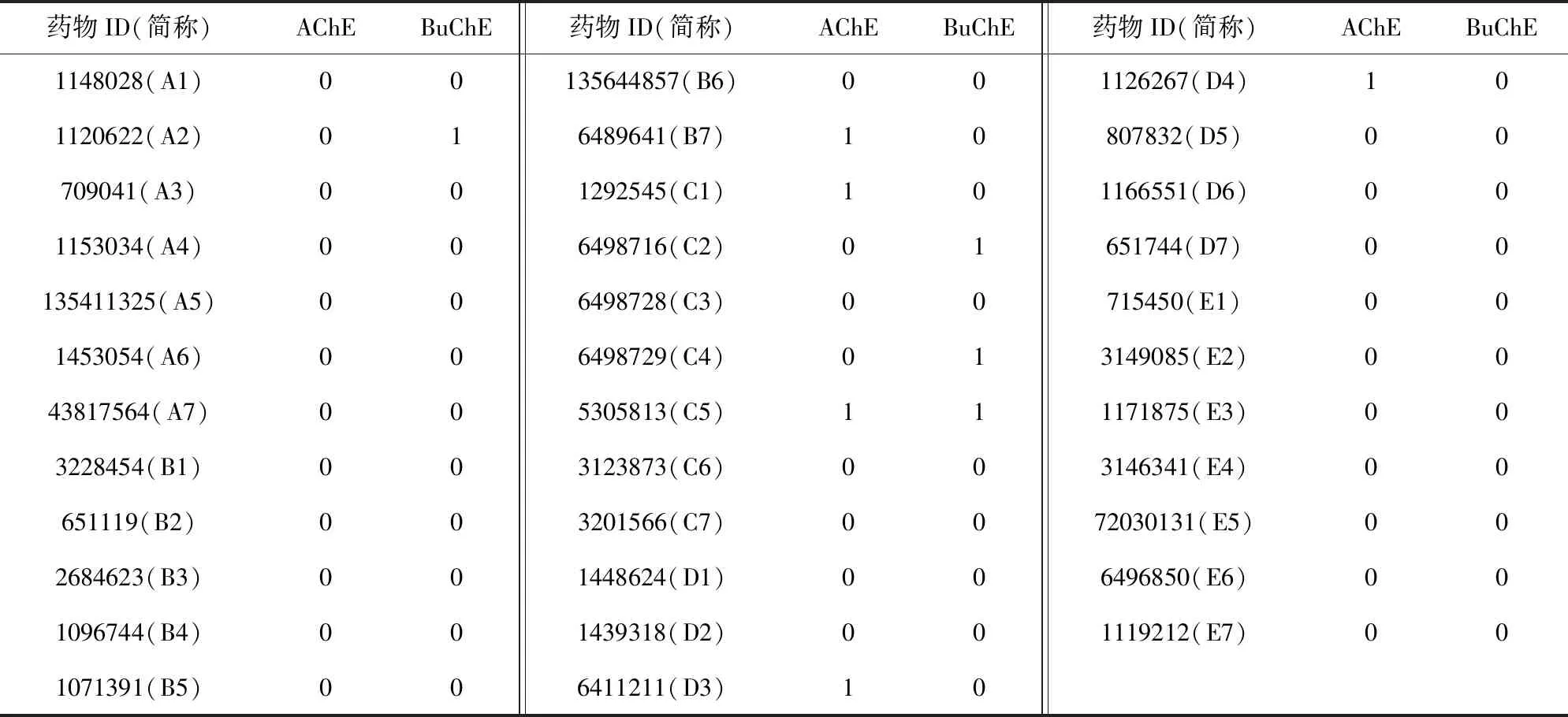
Table 9 The Manual Screening Drug Data Sets of AChE and BuChE Inhibition表9 手動篩選的AChE和BuChE抑制藥物數據集
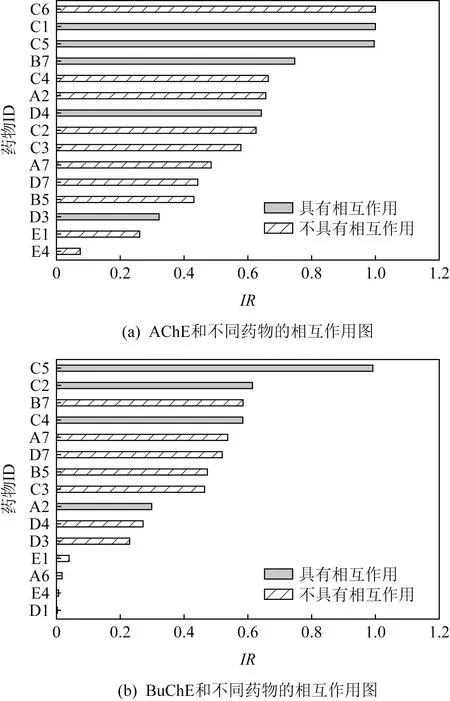
Fig. 8 The test results of protein-drug interaction圖8 蛋白藥物相互作用測試結果
由圖8可以看出,有相互作用的蛋白質藥物組合基本存在于預測的Top15范圍內,說明本文提供的模型具有實際的應用性.但不排除部分僅可抑制丁酰膽堿酯酶的藥物在預測和乙酰膽堿酯酶相互作用時成正相互作用,這在所難免.具備相似氨基酸序列的蛋白質,其與藥物相互作用的預測值也十分貼近,近似結構蛋白具備近似生物活性是預測PDI的重要理論支撐點但也是難以區分細節差異的原因,同時也是當前深度模型僅能做藥物篩選而不能做藥物設計的原因.想實現端到端模型設計藥物分子還需更深入的研究.
4 結 論
本文提出了一種基于自注意力機制和多藥物特征融合的蛋白質-藥物相互作用預測算法.首先,合理融合基于藥物分子結構特征的Morgan指紋、Mol2Vec表示向量以及消息傳遞網絡所提特征;隨后,將融合結果對由密集型卷積所提取的蛋白特征做注意力加權;最后綜合兩者特征,利用自注意力機制和雙向門控循環單元預測蛋白質藥物相互作用.本文在BindingDB,Kinase,Human,C.elegans這4個數據集上進行驗證,無論是與傳統機器學習算法相比還是與當前各深度學習算法相比,本文的算法都明顯更優.同時,本文根據所訓練模型針對抑制丁酰膽堿酯酶和乙酰膽堿酯酶做藥物篩選實驗.結果表明,本文所提模型具備可靠的應用性,但依然存在提升空間.首先,與其他基于深度學習的方法類似,黑盒模型依舊缺乏可解釋性[48].其次,利用同源蛋白以及相似結構藥物具備相似生物活性原理預測PDI和解決由細節差異帶來藥性影響的問題存有矛盾,當前還缺乏對此的深入研究.未來,會基于這2個問題繼續探索,不斷為藥物篩選工作提供新的解決方案.
作者貢獻聲明:華陽負責數據采集、實驗設計、編程實現和論文撰寫;李金星負責數據采集和實驗設計;馮振華參與論文修改;宋曉寧負責實驗指導,參與論文修改;孫俊參與論文修改;於東軍參與實驗指導和論文修改.
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
瘋狂英語·新策略(2019年10期)2019-12-13 08:43:28
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
當代陜西(2019年10期)2019-06-03 10:12:04
數學小靈通·3-4年級(2017年9期)2017-10-13 08:10:54
光學精密工程(2016年6期)2016-11-07 09:07:19
發明與創新(2016年38期)2016-08-22 03:02:52
