己二酰氯的綠色制備工藝
2022-08-31 08:51:20吳文靜戎有明周建成李乃旭
化工時刊 2022年8期
關鍵詞:催化劑
吳文靜 王 楠 戎有明 周建成, 李乃旭,*
(1. 東南大學 化學化工學院,江蘇 南京 211189; 2. 中微納米功能材料研究院有限公司,江蘇 南京 210044)
酰氯作為最活潑的酰基化試劑,可以與氨/胺反應生成酰胺,與醇反應生成酯,與羧酸根離子反應生成酸酐等[1]。其中己二酰氯作為有機中間體有多種用途,如己二酰氯與金剛石薄膜表面的官能團結合可得共價吸附層[2];已二胺和二酰氯之間進行界面縮聚是一種生產尼龍66的新方法[3];對稱二價分子被研究用于治療多種疾病[4],例如將己二酰氯參與反應合成的負載替莫唑胺(TMZ)的磁鐵礦三嵌段共聚物用于藥物遞送[5];近年來用己二酰氯制備的支化大分子或偶聯劑將典型的一維碳納米管結合到碳纖維表面形成“柔性—剛性”多尺度增強結構,是一種提高碳纖維增強復合材料界面性能的有效方法[6];己二酰氯與N,N′-己烷-1,6-二基-雙苯甲酰胺作用能夠產生可通過血腦屏障的毒蕈堿受體,這種受體可用于影響激動劑和拮抗劑與受體蛋白正構位點的結合[7];相比較于直接酯化的苛刻反應條件,使用己二酰氯可在相轉移催化條件下對單或雙取代酚進行酯化得到單酯或二酯,且獲得了更高的產物收率[8]。
酰氯的傳統制備方法中的亞硫酰氯法一般將N,N-二甲基甲酰胺(DMF)作催化劑與亞硫酰氯反應得到酰氯[9],亞硫酰氯沸點低,不易制備高沸點酰氯,且對設備腐蝕嚴重。三氯化磷法一般不需要結合催化劑[10],適合制備低沸點酰氯,但生成的亞磷酸易氧化成磷酸,具有腐蝕性,且受熱分解產生毒性氣體氧化磷。草酰氯法一般將其與DMF形成Vilsmeier中間體參與反應[11],原料為無色發煙液體,氣味刺鼻,反應過程劇烈不易控制。氰尿酰氯法的原料具有明顯刺激作用,破壞環境造成水體污染等[12,13]。這些方法均不符合綠色生產工藝標準。為解決酰氯化試劑毒性強、易揮發及溶劑回收困難等問題,本文采用三氯甲苯結合過渡金屬氯化物制備己二酰氯[14-16],同時聯產苯甲酰氯(如圖1),該合成路線簡單安全,為己二酰氯新工藝的開發以及應用提供了一定的借鑒價值。
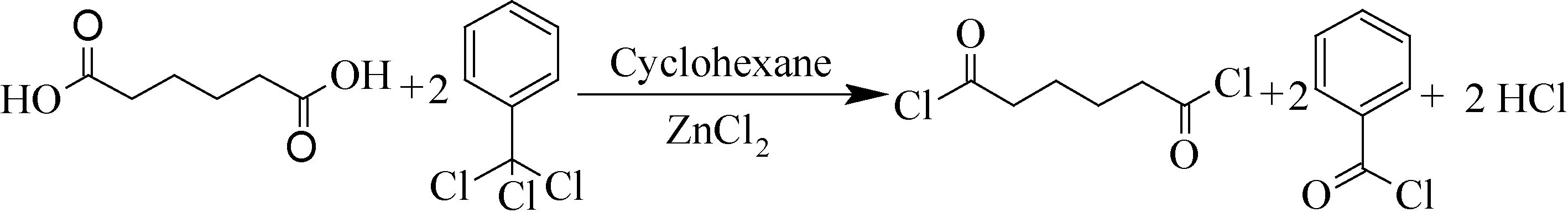
圖1 己二酰氯的制備
1 實驗部分
1.1 主要儀器與試劑
儀器:氣相色譜儀GC-9860(上海奇陽信息科技有限公司),傅里葉紅外光譜儀IR-408(日本島津公司),600 MHz核磁共振波譜儀AVANCE III HD(美國Bruker公司)。
試劑:己二酸,AR,上海易恩化學技術有限公司;環己烷,AR,上海易恩化學技術有限公司;三氯甲苯,99%,國藥集團化學試劑有限公司;四氫呋喃,≥99.8%,國藥集團化學試劑有限公司;甲苯,≥99.5%,國藥集團化學試劑有限公司;ZnCl2,AR,國藥集團化學試劑有限公司;AlCl3,AR,上海易恩化學技術有限公司;FeCl3,AR,上海易恩化學技術有限公司。
1.2 實驗方法
將一定量的己二酸、環己烷與三氯甲苯加入三口燒瓶中,攪拌,再稱取少量ZnCl2加入反應體系,30 ℃下反應3 h; 反應結束后于-0.05 MPa下旋蒸除去環己烷,減壓蒸餾后得到淡黃色澄清液體為己二酰氯。
1.3 結果與表征
己二酰氯的核磁氫譜見圖2,化學位移值δ1.57,1.25,0.88 ppm的小峰,峰高不大于1個質子,為雜質峰,其余2組主要峰的積分比為1∶1,對應兩個4重峰的圖譜中有2種質子。總積分值扣除雜質峰按8個質子分配,化學位移值δ2.94 ppm為鄰近羰基旁1、2號位H的信號峰,由于與羰基相連,p-π共軛,亞甲基氫化學位移值移向低場;化學位移值δ1.78 ppm為中間兩種3、4號位H的信號峰。1H NMR (600 MHz, CDCl3) 2.94 (tq, J=1.44,1.4 Hz), 1.97-1.17 (m)。
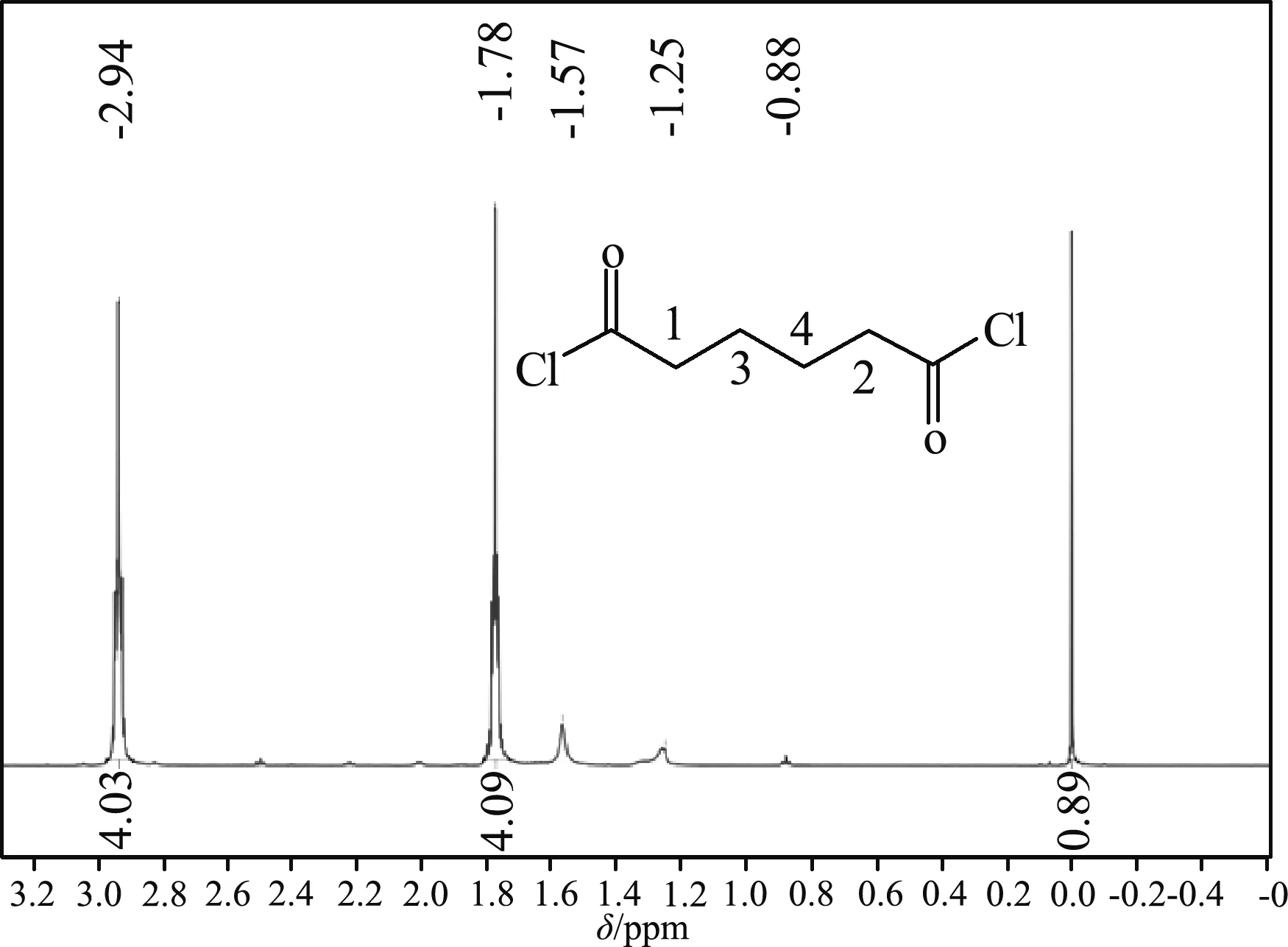
圖2 己二酰氯的核磁氫譜
己二酰氯的核磁碳譜見圖3,化學位移值δ173.10 ppm為連接雜原子的羰基中1、2號位C的信號峰,化學位移值δ23.57 ppm為不直接連接雜原子的脂肪鏈碳原子即3、4、5、6號位C的信號峰,δ46.21 ppm為C-Cl即同樣是1、2號位C的信號峰。
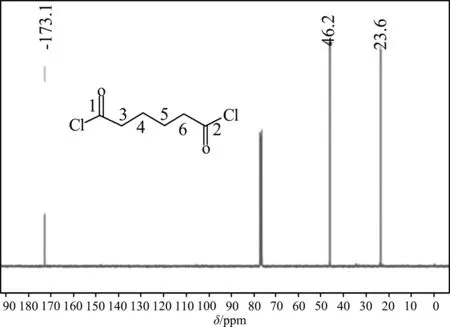
圖3 己二酰氯的核磁碳譜
己二酰氯的紅外譜圖見圖4,由圖4分析結構式的不飽和度為2,表明分子中有兩個C=C或C=O,3 300~2 800 cm-1區域為C-H伸縮振動,低于且接近3 000 cm-1的2 950 cm-1、2 877 cm-1一般為飽和C-H的伸縮振動吸收,1 805 cm-1和1 693 cm-1的峰屬于己二酰氯中脂肪酮的強伸縮振動吸收,1 407 cm-1的吸收峰表明己二酰氯中羰基伸縮振動峰,脂肪族C-Cl伸縮振動在850~550 cm-1區域有744 cm-1吸收峰,與產物的標準紅外光譜圖基本一致。

圖4 己二酰氯的紅外譜圖
2 單因素實驗
2.1 時間因素
在己二酸與三氯甲苯摩爾比為1∶2,27 mL環己烷,0.1 g ZnCl2,反應溫度30 ℃的條件下設置反應時間為3~7 h,己二酰氯收率與時間的關系如圖5。
由圖5可以看出,具有強親電能力的Lewis酸ZnCl2發生親電取代反應,前5 h收率逐漸上升,反應液中酸根離子與芐基正離子濃度逐漸增大,反應時間為5 h達最大值51.60%。5 h前消耗的酸根離子與己二酸產生配合物,但時間過短反應遠未達終點,體系中可能還有大量酸根離子及芐基正離子未參與到與己二酸形成中間體的步驟中。5 h后,體系副反應發生的概率增大。綜上,最適宜的反應時間為5 h。
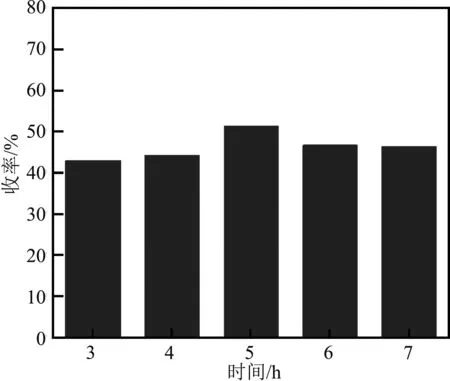
圖5 時間對收率的影響
2.2 溫度因素
己二酸與三氯甲苯摩爾比為1∶2,27 mL環己烷,0.1 g ZnCl2,設置反應溫度40 ℃~80 ℃,反應5 h。己二酰氯收率與溫度的關系如圖6。
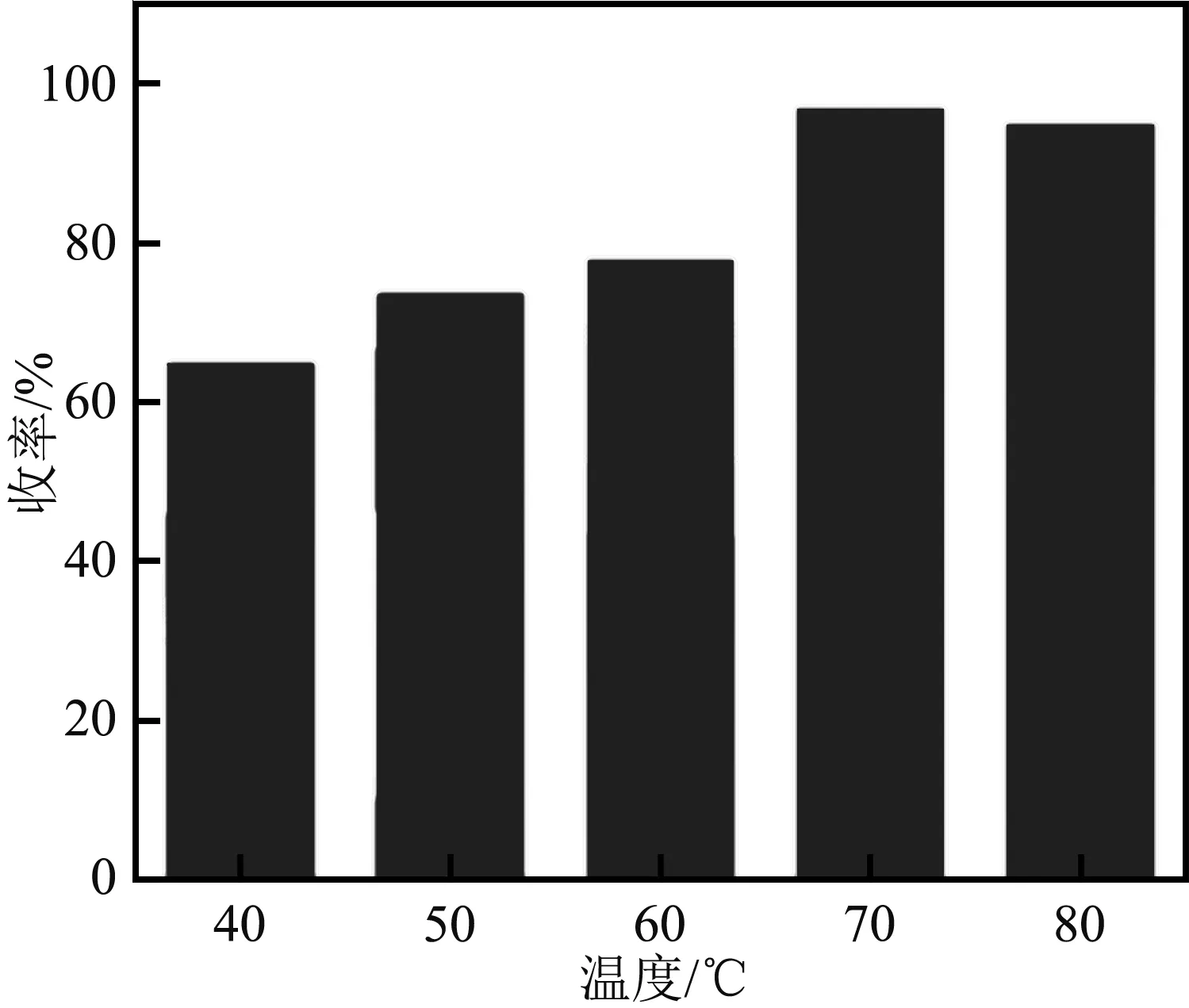
圖6 溫度對收率的影響
由圖6可知,40 ℃~70 ℃范圍內反應收率逐漸上升,在70 ℃達最大值96.88%,隨后下降。溫度作為宏觀量,高溫時分子熱運動劇烈導致反應速率增大,低溫時反應速率慢,同樣的反應時間內反應效率低,溫度由60 ℃到70 ℃時的收率提高幅度最大。溫度越高,內能越大,但同時伴隨溶劑汽化揮發,溫度過高,反應效果差。綜上,最適宜的反應溫度為70 ℃。
2.3 催化劑因素
己二酸與三氯甲苯摩爾比為1∶2,27 mL環己烷,0.1 g ZnCl2,70 ℃下反應5 h。選擇三種Lewis酸催化劑,分別是:分子晶體AlCl3、FeCl3及離子晶體ZnCl2,己二酰氯收率與催化劑種類的關系如圖7。
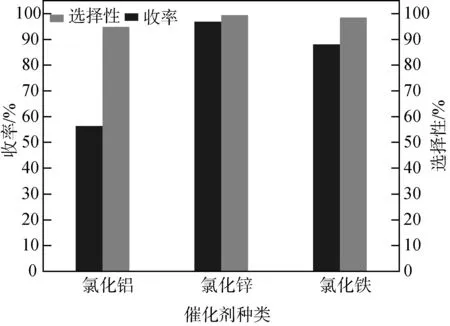
圖7 催化劑對收率的影響
實際使用過程中,FeCl3與AlCl3在常溫下具有強烈的吸水性,迅速吸收空氣中的水分并潮解,放出HCl形成酸霧,而酰氯需要在嚴格無水的環境下制備,所以這兩種催化劑制備條件相較具有強正負離子間靜電作用力的ZnCl2更加苛刻,三種催化劑使用過程中發現ZnCl2吸水性最弱,反應的加料量更易于控制。從圖7看出ZnCl2作催化劑時,收率最高。酸根離子與己二酸形成的配合物能夠被芐基正離子準確進攻且大部分用于活化配合物中的氧,其他位置幾乎不反應,反應完成后能夠分解重新還原成Lewis酸催化劑循環參與反應。綜上,最佳催化劑為ZnCl2。
2.4 溶劑因素
己二酸與三氯甲苯摩爾比為1∶2,0.1 g ZnCl2,70 ℃下反應5 h。選擇三種溶劑,分別是:甲苯、環己烷、四氫呋喃。己二酰氯收率與溶劑種類關系如圖8。
由于產物己二酰氯的極性弱,對比三種溶劑的極性大小,四氫呋喃極性最強,甲苯次之,環己烷極性最弱,根據相似相溶原理,環己烷與產物的極性差異度最小,從圖8明顯看出環己烷作為溶劑時,產物的收率最高,原料選擇性高。綜上,最佳溶劑為環己烷。
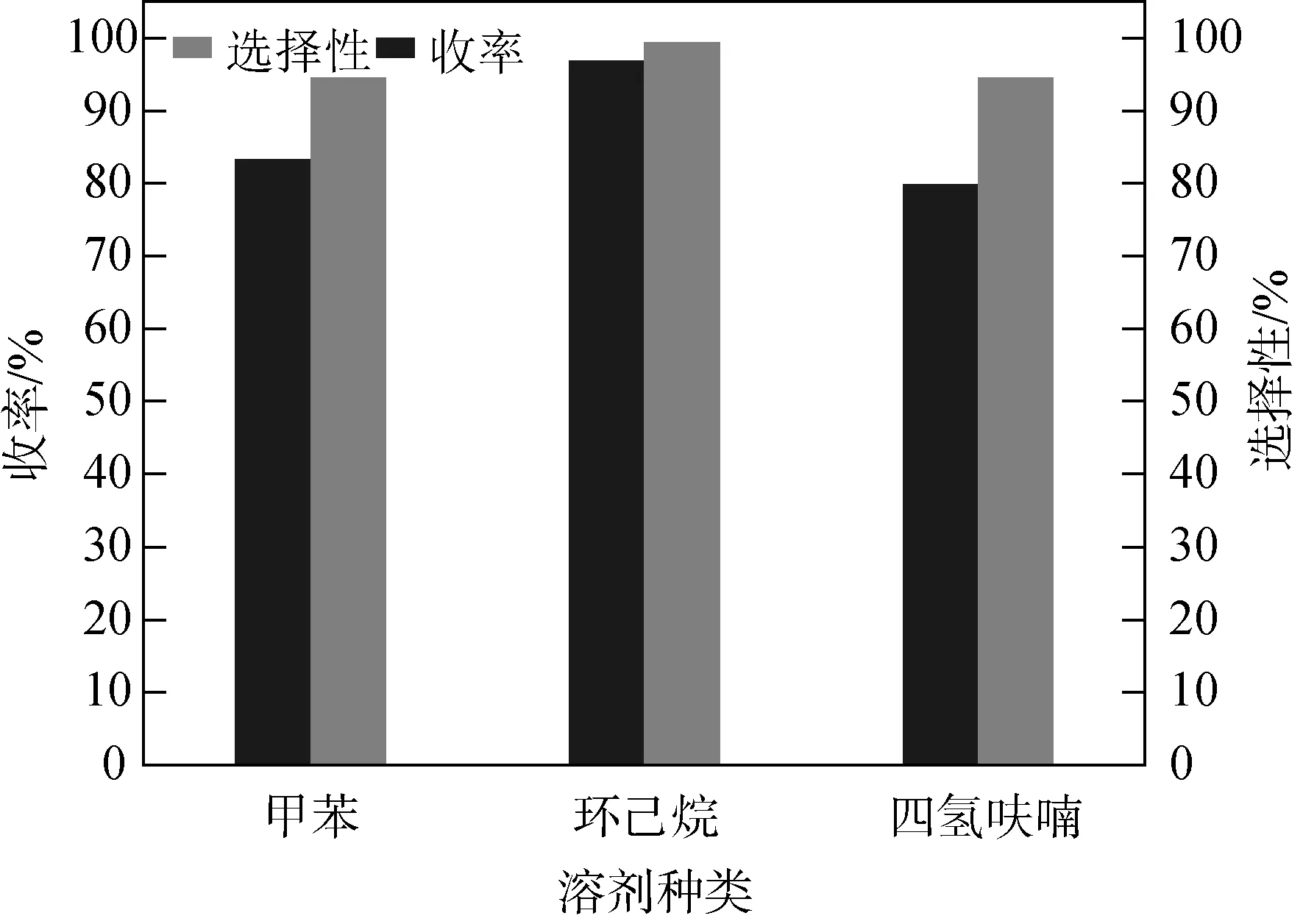
圖8 溶劑對收率的影響
3 反應機理
由于Lewis酸具有空軌道,吸電子能力強,在Lewis酸活化下,三氯甲苯中的氯易于離去,分別形成帶負電荷的酸根離子和帶正電荷的芐基離子;酸根離子活化己二酸中的活潑氫使其離去,二者再結合形成配合物;帶正電荷的芐基離子對配合物中的氧進行活化,使C-O鍵斷裂,形成C-Cl鍵,進而分解還原得到己二酰氯、苯甲酰氯和Lewis酸。反應機理如圖9。
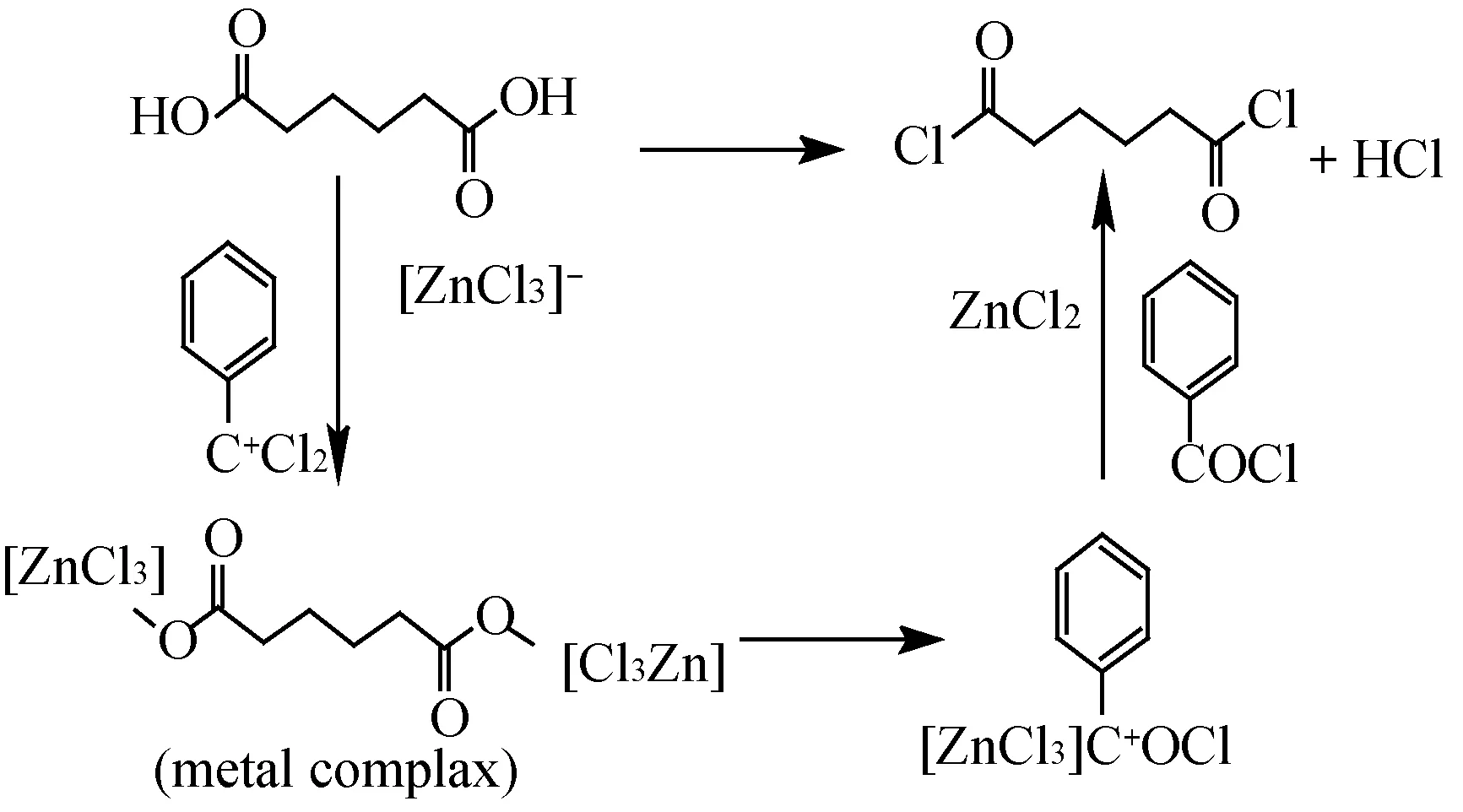
圖9 反應機理
4 結論
該路線以己二酸與三氯甲苯為起始底物,合成己二酰氯的同時聯產苯甲酰氯,經過單因素實驗確定時間為5 h,溫度為70 ℃,催化劑為ZnCl2以及溶劑為環己烷,最終以96.88%的收率得到目標產物己二酰氯。對目標產物進行核磁與紅外結構表征,均證實產物的基本結構。本文提出了一種制備己二酰氯的綠色合成新工藝,有效解決了使用傳統酰氯化試劑存在的毒性強、易揮發及溶劑回收困難等問題,且具有進一步擴大生產與開發應用的價值。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
