摻雜黑磷烯吸附鉻離子的第一性原理計算
2022-08-22 07:54:42張國英管永翔
沈陽師范大學學報(自然科學版) 2022年2期
關鍵詞:體系
張國英, 管永翔, 于 樂
(沈陽師范大學 物理科學與技術學院, 沈陽 110034)
0 引 言
自工業革命以來,人類在創造財富的同時也嚴重污染了人們賴以生存的生態環境。水體作為人類生存和發展的重要物質也遭受了不同有害物質的污染,如鉻、鎘、鎳、砷等有害離子不僅會降低水質使其惡化,還會被生物攝食吸收、富集,通過食物鏈逐級放大,一旦被人體吸收會嚴重影響人類生理活動,造成不可逆的身體傷害,甚至死亡。鉻離子被攝入人體會侵害人體神經,造成腦損傷,而且會誘發癌癥。因此,從環保健康的角度來說,開發出吸附力強的吸附材料來消除污水和廢水中的鉻離子是至關重要的[1-4]。石墨烯是近年來炙手可熱的研究對象,較大的比表面積使其具有良好的吸附性能,在吸附領域有著廣泛的應用前景[5-7]。譚心等[8]運用在石墨烯表面增加雙空位缺陷的方法,增強了堿金屬原子在石墨烯表面的吸附能力。由于石墨烯的出現,研究者們將目光放在了二維材料領域,試圖從新材料里尋求突破[9]。黑磷(BP)烯是磷單質的一種同素異構體,與石墨烯有著類似的二維層狀結構,在黑磷烯單原子層中,每個磷原子同相鄰的3個磷原子以共價鍵形式相連,形成蜂窩狀的褶皺結構[10-12],正是這種特殊的結構,使它呈現出優于其他二維材料的獨特性質和應用前景[13-15]。Li等[16]研究了超高靈敏度的懸浮原子薄層黑磷傳感器,實現了汞離子的快速無痕跡痕量檢測,檢測限為0.01PPb。Liu等[17]研究了一種用于增強光-物質界面和化學傳感的集成黑磷(BP)傾斜光纖光柵(TFG)配置,通過一種用于BP納米片沉積的原位逐層技術,實現了在特定纖維柱表面上的高質量BP涂層,BP-TFG用于檢測重金屬鉛離子,表現出高達0.5×10-3dB/PPb的超高靈敏度。雖然黑磷烯已經被研究,并且成果豐碩,但是在吸附領域還存在許多懸而未決的問題[18]。
本文研究了純凈黑磷烯與摻雜黑磷烯體系對鉻離子的吸附作用,通過對比吸附能、共價鍵鍵級、態密度等實驗數據,分析黑磷烯能否作為除去鉻離子的吸附材料,并通過摻雜進一步提升其吸附能力。該結果對于水中去除金屬離子具有重要意義,同時也進一步拓展了黑磷烯的實際應用范圍。
1 計算方法與模型
計算基于密度泛函理論的第一性原理方法,運用CASTEP軟件包,交換關聯能采用廣義梯度近似,對黑磷烯進行參數計算和結構優化[19]。在布里淵區積分計算時,基于黑磷體材料的計算結果,通過與黑磷體材料的實驗值[20]相比發現,K網格設置為3×3×3,截斷能設置為330 eV比較合理。為了節省計算資源,選取2×2×1的單層黑磷烯的超原胞結構,原胞共16個磷原子,同時為了防止BP烯層間相互干擾,在C方向上建立了真空層。優化2×2×1的單層黑磷烯超原胞后,得到晶格常數a=3.38 ?,b=10.56 ?,c=4.21 ?,均與已發表的結論相吻合[21]。
為了更加直觀地對比鉻離子在不同黑磷烯體系下的吸附能力,定義式(1)為鉻離子的吸附能表達式[22]:
Ead=(EBPsheet+ECr2+)-E(Cr2+/BPsheet)
(1)
式中:ECr2+表示單個鉻離子的能量;EBPsheet表示摻雜或未摻雜BP烯的能量;E(Cr2+/BPsheet)表示鉻離子吸附后整個體系的能量。
形成穩定吸附后,BP烯與鉻離子間產生了電荷轉移,定義式(2)為鉻離子與BP烯之間轉移的電荷量[23]:
ΔQ=|Qad-Q|
(2)
式中:ΔQ表示體系之間電荷的轉移數;Qad表示鉻離子吸附后所帶電荷數;Q表示鉻離子吸附前所帶電荷數。
2 結果與討論
2.1 結構性質
摻雜其他原子不僅會改變黑磷烯原有的結構,而且會影響黑磷烯對于鉻離子的吸附性能。為了確定鉻離子在純凈黑磷烯上最穩定的吸附位置,考慮了3種吸附位置:頂位(T位),即鉻離子位于磷原子(P原子)頂部正上方位置;橋位(B位),即鉻離子位于黑磷烯2個P原子成鍵中間的上方位置;空位(H位),即鉻離子位于相鄰3個P原子之間的中空位置的上方。優化純凈BP烯3種吸附結構,結果發現,Cr2+吸附于空位(H位)時最穩定。圖1給出了黑磷烯吸附Cr2+后的優化結構。繪圖時設定當鍵長小于3 ?時,圖中顯示成鍵。此時Cr2+與相鄰4個P原子成鍵,即4配位。但2個原子間是否真正形成了共價鍵,后面通過鍵級的計算進行了分析,結果發現,Cr2+與P1并沒有形成共價鍵,所以嚴格地講,Cr2+與相鄰3個P原子成鍵,即3配位。依據公式(1),計算得Cr2+的吸附能為12.472 eV。吸附能為正值說明Cr2+的吸附過程是放熱過程,可以自發進行,此結構下的鍵長及吸附能見表1。
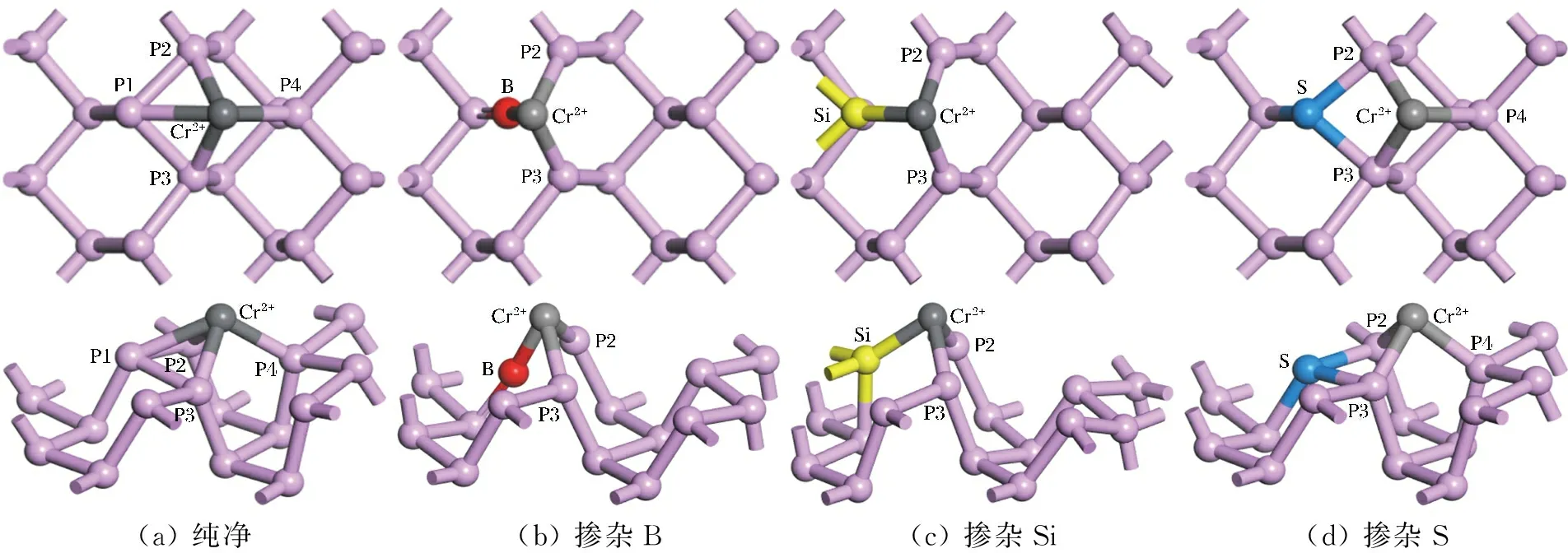
(a) 純凈(b) 摻雜B(c) 摻雜Si(d) 摻雜S

表1 純凈、B摻雜、Si摻雜、S摻雜時BP烯吸附Cr2+的優化結構參數Table 1 Structural parameters of Cr2+ adsorbed on pure, B doped, Si doped and S doped black phosphorene
同理,摻雜硼(B)、硅(Si)、硫(S)時也考慮3種吸附位置。幾何優化后結果顯示,3種不同摻雜體系中,Cr2+均吸附在褶皺六邊形頂部的空心位置上方,如圖1所示。不同的是,B摻雜體系中,Cr2+與相鄰的2個P原子和1個B原子成鍵;Si摻雜體系中,Cr2+與相鄰的2個P原子和1個Si原子成鍵。這種成鍵方式的原因可能是原子外圍電子排布不同或者原子半徑不同,導致B原子與Si原子均不與鄰近的P2和P3原子成鍵(圖1(b),(c));S摻雜體系中,Cr2+與鄰近3個P原子成鍵,即三配位。根據公式(1)計算得出,B摻雜、Si摻雜、S摻雜3種體系下Cr2+的吸附能分別是11.741 eV,12.529 eV,13.336 eV,吸附能數值均為正值表明3種摻雜體系下的吸附過程都是放熱過程,可以自發進行。通過與純凈黑磷烯吸附能的對比發現,Si摻雜和S摻雜可以有效提高吸附能,其中S摻雜提升效果較為明顯,提升了0.864 eV。
2.2 BP烯吸附Cr2+的電子性質
本文通過對純凈、B摻雜、Si摻雜和S摻雜4種體系吸附Cr2+的態密度計算,繪制了4種BP烯體系在吸附Cr2+前后的總態密度圖及吸附Cr2+前后各原子的局部態密度圖,用來進一步研究BP烯吸附Cr2+的電子及成鍵性質。為了更清楚地顯示不同原子的態密度,繪制時高斯展寬設置為0.05 eV,如圖2所示,費米能級處于能量0點處。圖2(a)最上面標志(—BP)的圖為純凈黑磷烯吸附前的總態密度,可見費米能級處于價帶頂,其帶隙值約為1.5 eV。在吸附Cr2+后,見圖2(c)最上面標志(—BP(+Cr))的小圖,從吸附前帶隙值1.5 eV到吸附后帶隙值變為0,說明Cr2+吸附使體系的導電性大大增強。比較圖2(c)和圖2(d )最上面的2個小圖,可以看到費米能級以下的成鍵態是Cr2+和P共同貢獻的,可見Cr2+和P間存在共價作用。B摻雜BP烯體系在吸附Cr2+前,如圖2(a)第2欄標志(—BP(+B))的小圖,由于B原子的存在,在0.8~1.3 eV形成了雜質能帶。由圖2(b)第2欄標志(—P,—B)的小圖可以看出,雜質能帶主要是由P原子貢獻的,B也有一定的貢獻。靠近導帶底的B摻雜引起的雜質帶提高了導電性。在吸附Cr2+后,見圖2(c)第2欄標志(—BP(+B+Cr)的小圖,可見產生不止1個雜質帶。結合圖2(d)第2欄標志(—P,—Cr,—B)的小圖可知,能隙中央的雜質帶主要是由Cr2+和P原子貢獻的,較高的雜質帶是由B和Cr2+貢獻的。雜質帶的存在會提高體系的導電性。仔細觀察圖2(d)第2欄標志(—P,—Cr,—B)的小圖,Cr2+與P和B的耦合較小(相比于圖2(d)第1欄Cr2+和P的耦合),說明B摻雜使Cr2+與黑磷烯表面間的共價作用減弱。對于Si摻雜,見圖2(a)第3欄標志(—BP(+Si))的小圖,吸附Cr2+前,Si原子摻雜在價帶頂附近形成了雜質能帶,此時帶隙為1.0 eV左右,可見Si摻雜同樣能改善黑磷烯的導電性。結合圖2(b)第3欄標志(—P,—Si)的小圖可知,能隙價帶頂附近的雜質帶主要是由Si和P原子貢獻的。吸附Cr2+后,見圖2(c)第3欄標志(—BP(+Si+Cr))的小圖,雜質帶幾乎充滿,可見吸附后體系導電性大大提高。仔細觀察圖2(d)第3欄標志(—P,—Cr, —Si)的小圖,可見0 eV附近的雜質帶是由Cr2+與P原子貢獻的;較高的雜質帶主要是由Cr2+與Si原子貢獻的,Cr2+與P原子也有一定的貢獻。此外,還可以看到,在費米能級以下的成鍵態中,Cr2+與P在0 eV附近的耦合比Cr2+與Si的強,可見Cr2+與P的共價作用比Cr2+與Si的強。最后討論S摻雜,如圖2(a)最下面一欄標志(—BP(+S))的小圖,在導帶底附近有一雜質能帶,這主要是S原子與P原子共同作用的結果。從圖2(b)最下面一欄標志(—P,—S)的小圖可以看出,費米能級接近導帶底,說明S摻雜使黑磷烯變成了n型半導體。在吸附Cr2+后,見圖2(c)最下面一欄標志(—BP(+S+Cr))的小圖,費米能級處于價帶頂,說明Cr2+使S摻雜的n型BP烯又變回了p型半導體。雜質產生的能帶與導帶相連,使帶隙變為0.7 eV左右,可見導電能力還是很強。仔細觀察圖2(d)最下面一欄標志(—P,—Cr,—S)的小圖,可見成鍵態由Cr2+,P,S共同貢獻。由前面的優化結果可知,Cr2+不與S成鍵(間距大于3 ?),所以此時Cr2+與P形成較強的共價鍵。
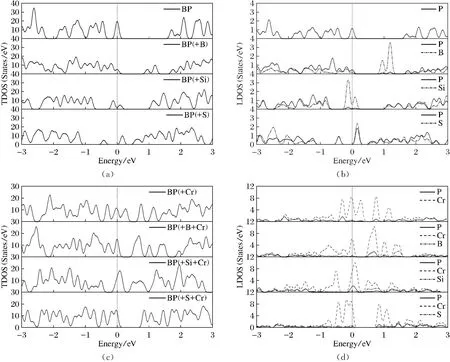
(a)(b)(c)(d)
2.3 BP烯吸附Cr2+成鍵性分析
離子鍵是通過2個或多個原子失去或獲得電子而成為離子后形成的。在BP烯吸附Cr2+的過程中,Cr2+與BP烯表面P原子或摻雜原子發生電荷轉移,電荷量轉移的多少標志著離子鍵的強弱,電荷量轉移越多說明體系內所形成的離子鍵越強,也表示此狀態的BP體系對Cr2+的吸附力越強。從表2可以看出,S摻雜BP烯電荷轉移為1.52 e,是4種BP烯體系中電荷量轉移最多的,其離子鍵作用是4種體系中最強的,說明S原子增強了BP烯表面失去電子的能力。Si摻雜電荷轉移為1.48 e,僅次于S摻雜。而B摻雜電荷轉移量比純凈BP烯少,說明從離子鍵強度方面討論,B摻雜不能提升BP烯對Cr2+的吸附能力。
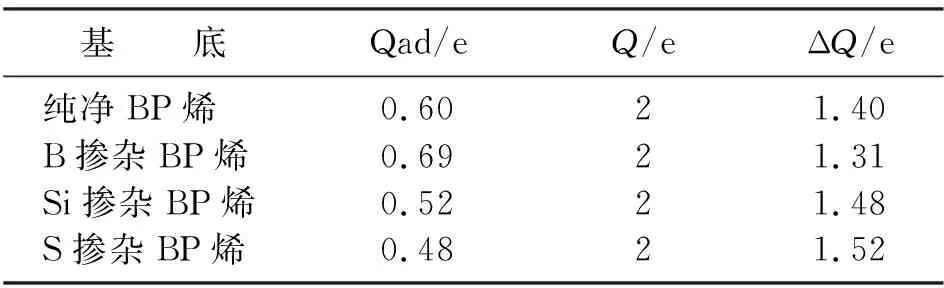
表2 Cr2+失去電荷情況Table 2 The charge loss of Cr2+
在吸附過程中,Cr2+與P原子或摻雜原子發生電荷轉移的同時,也會與其形成共價鍵作用,共價鍵也是一種強作用力,可以通過鍵級的大小來判斷其強弱。鍵級是表示相鄰的2個原子電子云的交疊程度,鍵級愈大,共價作用越強。表3給出了4種BP吸附體系Cr2+與成鍵原子的鍵級。純凈BP烯吸附的Cr2+與相鄰4個P原子成鍵(鍵長小于3 ?),其中Cr2+與P1的鍵級為負值,說明二者之間沒有電子云交疊,可見沒形成共價鍵。嚴格地說,Cr2+與相鄰3個P原子成鍵,是三配位的。B摻雜BP烯,Cr2+與相鄰2個P原子及B原子成鍵,鍵級為0.31,0.32和0.41,通過與純凈BP烯對比,發現從共價鍵強弱角度分析,B摻雜未能提升BP烯的吸附能力。Si摻雜BP烯,Cr2+與相鄰2個P原子及Si原子成鍵,鍵級分別為0.48,0.47和0.43,鍵級數值總體比純凈BP烯Cr2+與3個P原子間的鍵級大,可見共價作用增強。S摻雜BP烯,Cr2+與相鄰3個P原子成鍵,鍵級分別為0.48,0.48和0.47,鍵級數值總體上是4種BP烯吸附體系中最大的,即摻雜S后,Cr2+與黑磷烯表面共價作用最強。結合上面離子鍵討論的結果,說明從成鍵角度分析,S摻雜可以有效提高BP烯對Cr2+的吸附能力。

表3 Cr2+吸附于純凈和摻雜BP烯后的共價鍵鍵級Table 3 Covalent bond order of Cr2+ adsorbed on pure and doped black phosphorene
3 結 論
本文研究了Cr2+在純凈、B摻雜、Si摻雜和S摻雜4種不同BP烯體系的吸附行為,結果發現,Cr2+穩定吸附時,Cr2+在純凈、B摻雜、Si摻雜和S摻雜BP烯體系中均是三配位。Si和S摻雜BP烯吸附Cr2+時的吸附能較純凈黑磷烯有較大提高,說明Si和S摻雜有效地提高了BP烯對Cr2+的吸附能力。根據態密度的計算結果可以看出,摻雜調制了BP烯及其Cr2+吸附體系的導電性,同時摻雜也影響了Cr2+與黑磷烯表面的共價鍵作用。通過電荷轉移和鍵級的計算結果得出,S和Si摻雜增強了BP烯表面原子與Cr2+間的離子鍵和共價鍵作用,但B摻雜卻使此作用減弱。綜上,S摻雜可以改善BP烯對Cr2+的吸附能力,S摻雜BP烯可以作為去除Cr2+的吸附材料。
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
