阿苯達唑納米混懸劑的研制及其評價
2022-08-11 07:06:22謝書宇趙寶凱
中國獸藥雜志 2022年7期
關鍵詞:血漿
田 凱,許 丹,盧 迪,謝書宇,趙寶凱*
(1.沈陽偉嘉生物技術有限公司.沈陽110020;2.華中農業大學,武漢.430070)
阿苯達唑(albendazole,ABZ),別名丙硫咪唑,化學名稱為5-丙硫基-1H-苯并咪唑-2-氨基甲酸甲酯,是苯并咪唑類的廣譜抗寄生蟲藥,現已被世界衛生組織(WHO)認定成為抗棘球蚴病的首要藥物之一。近年來臨床應用廣泛,且療效得到了廣泛認可[1]。遺憾的是,阿苯達唑溶解性低、首過效應強,在胃腸道吸收率極低,在血漿和肝臟組織中濃度不高,對非腸道寄生蟲如細粒棘球蚴病等治愈率僅為30%[2]。目前市場上阿苯達唑經常使用的制劑類型以片劑、粉劑和混懸劑為主。混懸劑在給藥途徑上優于片劑和粉劑,但由于阿苯達唑溶解度較差,普通混懸液的生物利用度依然很低。目前阿苯達唑的溶解性低、生物利用度低等缺點已經嚴重制約其在獸醫臨床上的使用,因此開發新制劑技術提高其生物利用度非常必要。
納米晶體(Nanocrystals,NC),是將水難溶性的藥物或藥物化合物在穩定劑的作用下分散于介質中(常以水為介質),利用機械研磨、高壓均質、控制析晶等納米化工藝,將藥物粒徑降低至 1 μm以下,使藥物形成納米膠體分散體系,可以明顯使難溶性藥物的溶解度及生物利用度得以改善。相比其他解決藥物難溶性問題的方法,此方法不需要加入任何載體,只需在制備過程中加入穩定劑來穩定制備的納米晶體,具有生產工藝簡單、納米混懸液制劑溶解性好、生物利用度高的優點,因此利用此方法可有效解決難溶性藥物臨床應用問題,有利于藥物在腸道內吸收并延長藥物的體內作用時間,從而提高難溶性藥物的生物利用度[3-4]。
通過改變傳統的混懸劑制備工藝,本研究利用反溶劑法-高壓勻質法制備阿苯達唑納米混懸劑,并考察該制劑的藥劑學特征及穩定性;同時進行大鼠體內的藥代動力學研究,考察其生物利用度,為提高阿苯達唑的臨床效果提供新的策略。
1 材 料
1.1 藥品與試劑 阿苯達唑對照品(含量99.9%,批號100373-201103),甲苯咪唑對照品(含量99.8%,批號H1032011),均購自中國獸醫藥品監察所;阿苯達唑亞砜對照品(含量99.07%,批號G119446),購自德國Dr.Ehrenstorfer GmbH公司;阿苯達唑原料(含量99.2%,批號9011806059),購自連云港亞暉醫藥有限公司;蘋果酸,購自西安天正藥用輔料有限公司;肝素鈉、吐溫80、苯甲酸鈉均購自國藥集團化學試劑有限公司;聚維酮K30(PVPK-30)(批號20180603),購自湖州展望藥業有限公司。阿苯達唑伊維菌素粉(含量10%,規格250 g,批號2019040101),沈陽偉嘉生物技術有限公司;阿苯達唑混懸液(含量10%,規格100 ml,批號2019101701),佛山市正典生物技術有限公司。甲醇、乙腈,色譜純;氫氧化鈉、乙酸乙酯、冰醋酸、95%乙醇,分析純;均購自國藥集團化學試劑有限公司。
1.2 試驗儀器 LC-1260高效液相色譜儀,美國AgiLent公司;分析天平MS105DU/A,梅特勒-托利多公司;PGC-01D型氮吹儀,天津艾維歐科技發展有限公司;UV-2501PC 紫外分光光度計,日本島津公司;PHS-3C型pH計,上海儀電科學儀器股份有限公司;TGL-18C高速臺式離心機,上海安亭科學儀器廠;XH-C渦旋混合器,常州邁科諾儀器有限公司;XHF-DY高速分散器,寧波新芝生物科技股份有限公司;Zetasizer Nano-ZS90納米粒度電位儀,英國馬爾文儀器有限公司;高壓勻質機APV2000,德國APV公司。
1.3 試驗動物 清潔級 SD大鼠30只,雄性,體重200±20 g,由遼寧長生生物技術股份有限公司提供。試驗前正常飼養1周,自由飲水和采食;并于試驗前1 d檢查確認試驗動物處于良好的健康狀態。
2 方 法
2.1 阿苯達唑納米混懸劑的處方篩選 根據《獸藥質量標準》(2017版)收載,阿苯達唑混懸液規格為100 mL:10 g。故本研究以阿苯達唑為主藥,按照混懸劑制備要求,通過單因素試驗進行納米混懸劑中助溶劑、穩定劑的篩選[5]。通過粒徑測定,最終確定L-蘋果酸為助溶劑,吐溫80 與PVPK-30為穩定劑,苯甲酸鈉為防腐劑。
2.2 制備工藝 按照配方比例,先將助溶劑蘋果酸用少量蒸餾水溶解,加熱至80 ℃~90 ℃,將阿苯達唑原料加入其中,保持加熱溫度使其完全溶解于助溶劑溶液中;另稱取穩定劑吐溫80和PVPK-30溶于適量蒸餾水中,使用高速分散器在剪切條件下將溶解的阿苯達唑溶液緩慢加入穩定劑溶液中,使阿苯達唑重新結晶析出,通過剪切獲得較小的粒徑,得到制劑初混液。將制劑初混液置于高壓均質機中,設定溫度、壓力及均質次數進行均質,即獲得納米混懸液。向制得的納米晶體混懸液中加入適量已溶解的防腐劑,攪拌均勻并調節pH,最后加純化水至全量,混合均勻即得。
2.3 納米混懸劑的質量評價
2.3.1 納米混懸劑的質量檢測 參照《獸藥質量標準》(2017版)中“阿苯達唑混懸液”的質量標準進行檢查。檢測項包括制劑的外觀、色澤、pH值、沉降體積比及含量。重分散性檢測項參照《藥劑學》(第七版)中混懸劑“重新分散性”檢測方法進行檢查。
2.3.2 納米顆粒形態及粒徑分布 將納米混懸液進行掃描電鏡檢測,觀察藥物形態特征。并測定納米混懸液的粒徑大小及分布。
2.3.3 制劑穩定性研究 參照《中華人民共和國獸藥典》(2015版)中的“制劑穩定性試驗指導原則”,對該納米混懸劑進行常溫、加速考察試驗,定期6個月,研究該制劑在高溫、高濕等條件下的穩定性。
2.4 藥物動力學試驗
2.4.1 藥物動力學試驗設計 將大鼠隨機分為三組,一組給藥阿苯達唑伊維菌素粉,一組給藥本研究制備的阿苯達唑納米混懸液,一組給藥佛山正典的阿苯達唑混懸液,給藥前12 h禁食,自由飲水,給藥后6 h恢復進食。根據阿苯達唑在豬上的臨床使用劑量,并參考相關文獻[6],進行換算得出大鼠的使用劑量為45 mg/kg.bw,進行灌胃給藥;三組藥物給藥前均用適量水稀釋制成儲備液備用,對每只大鼠進行稱重標記,按照 2 mL/0.2 kg 大鼠體重進行灌胃。并于給藥前、給藥后0.5、1.5、3、4、5、6、7、8、12和24 h從尾靜脈取血約0.5 mL置于肝素抗凝的離心管中,4000 r·min-1離心10 min,分離血漿,保存于-20 ℃,待測定。
2.4.2 大鼠血中阿苯達唑亞砜(ABZSX)的色譜條件 色譜柱:Ultimate?XB-C18,3 μm,4.6×250 mm;流動相(甲醇:乙腈 =1∶1)與水按照70∶30(V/V)的初始比例進行梯度洗脫,在 0~34 min水相比例由 70%→30%;在 34~40 min其比例由 30% →70%;檢測波長 295 nm;流速1.0 mL·min-1;柱溫 35±1 ℃[2]。
2.4.3 血漿樣品處理 精確量取200 μL大鼠血漿,置于2 mL離心管中,依次加入 NaOH溶液50 μL(0.4 mol/L),甲苯咪唑(MBZ)內標溶液 100 μL(10 μg/mL)及1.0 mL乙酸乙酯,渦旋振蕩 3 min,于12000 r·min-1離心10 min;吸取上清液置于另一離心管,并于第一管中再加入0.5 m L乙酸乙酯,重復前述操作進行渦旋振蕩及離心,合并兩次上清液,40 ℃氮氣流吹干,殘留物用200 μL甲醇乙腈混合液(V/V=1∶1)溶解,12000 r·min-1離心 10 min,取上清即為待測樣品,進樣量50 μL[2,7]。
2.4.4 血漿標準曲線的建立 精密量取空白血漿200 μL 9份,分別置 2 mL離心管中,留一支作為空白對照,其他分別加入等量的甲苯咪唑(MBZ)內標溶液,然后各加入不同量的阿苯達唑亞砜(ABZSX)標準工作液,得到藥液濃度依次為0.01、0.02、0.05、0.2、1、3.5、7、10 μg/mL的標準血漿樣品。按照“血漿樣品處理”方法處理,進行HPLC分析,記錄色譜圖。以測得的ABZSX峰面積與內標MBZ峰面積的比值為橫坐標(x),ABZSX濃度(y)為縱坐標,建立血漿中ABZSX標準曲線,擬合回歸方程,求得相關系數(r)。
2.4.5 方法學驗證 取空白血漿200u L,分別加入經稀釋的高、中、低三個濃度的ABZSX標準工作液,混合均勻,以制成含ABZSX濃度分別是0.2、3.5、10 μg/mL的血漿樣品,按照“血漿樣品處理”方法處理,進行HPLC檢測,每個濃度一天內制備5個平行樣品檢測,考察日內變異系數;連續重復檢測5 d,測定日間變異系數。并計算方法的回收率、準確度及精密度。
2.4.6 大鼠血漿中藥物濃度的測定 同一只鼠的血漿樣品在同一分析批內完成,按照“血漿樣品處理”方法處理檢測,將得到的ABZSX的峰面積代入標準曲線回歸方程,計算各時間點血漿中ABZSX的濃度。
2.5 數據分析 利用ExceL軟件分析處理檢測方法學數據,繪制血漿標準工作曲線及藥-時曲線圖;并利用藥動學分析軟件Winnonlin5.2以非房室模型處理血漿濃度-時間數據,獲得的藥動學參數以平均數±標準差(x±s)表示。
3 結果與分析
3.1 阿苯達唑納米混懸劑的制備工藝 以納米混懸液粒徑為評價指標,根據配方篩選單因素試驗結果,選擇L9(34)表設計,進行正交試驗,進一步優化勻質壓力(A)、均質次數(B)、勻質溫度(C)的工藝條件。正交試驗各因素與水平、正交設計試驗結果見表1,表2。
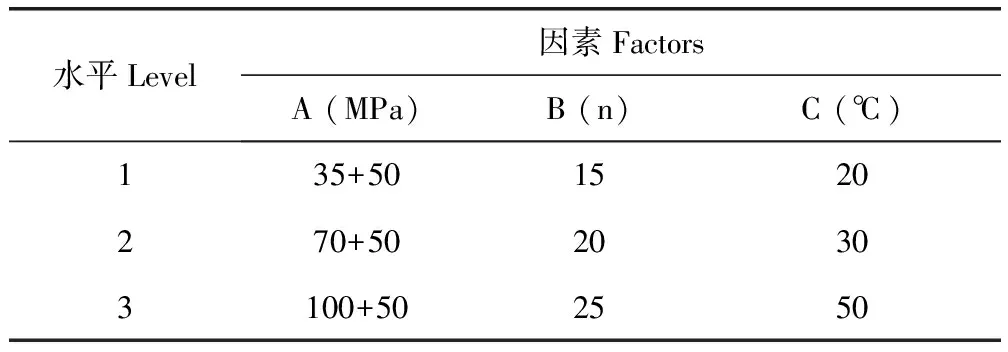
表1 正交設計試驗的因素與水平
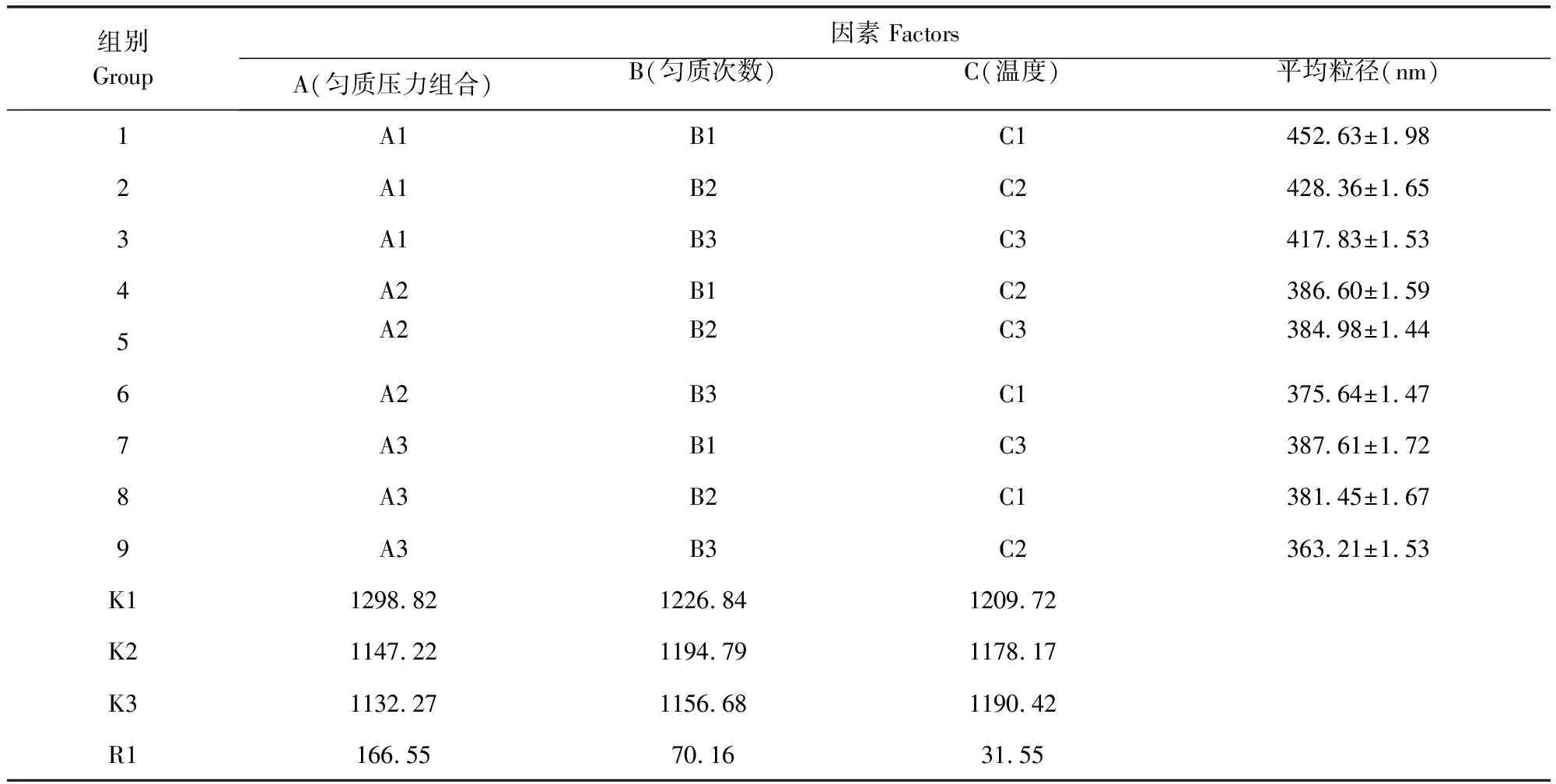
表2 正交設計表
納米晶體粒徑受各因素影響的主次關系為A>B>C,說明勻質壓力對納米晶體制備影響最大。根據正交試驗結果,優選的工藝參數為溫度30 ℃,100 MPa和50 MPa下各循環25次。同時為保證制備工藝的可行性,按照最佳工藝條件進行3次不同批量的重復試驗,所得結果平均粒徑為(365.43±1.68)nm,說明優選出的制備工藝穩定可行。
3.2 阿苯達唑納米混懸液的質量評價
3.2.1 質量檢測結果 該納米混懸劑外觀為類白色,長時間靜置,性狀無明顯變化。pH值在5.0~7.0內,含量為99.6%,沉降體積比為0.99,符合規定。
室溫下,將該制劑置于100 mL 具塞量筒中,密塞放置,沉降7 d后,并以20 次/min 的速度翻轉量筒,底部沉降物隨即消失,說明制劑重分散性較好。
3.2.2 形態觀察與粒徑分布結果 納米混懸液在掃描電鏡下藥物形狀大小均一,且中位粒徑為358.1 nm。具體見圖1、表3。

表3 阿苯達唑納米混懸液的粒徑分布
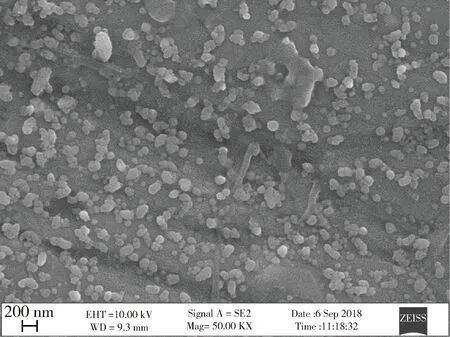
圖1 阿苯達唑納米混懸液掃描電鏡圖片
3.2.3 穩定性考察結果 將納米混懸劑在溫度30±2 ℃,相對濕度65±5%的條件下進行加速試驗,并分別于1個月、2個月、3個月、6個月考察樣品的各檢測項,與常溫放置樣品比較均無明顯差異。同時對放置6個月的樣品進行粒徑檢測,結果顯示粒徑有所增加,但仍為納米級,具體結果見表4、表5。
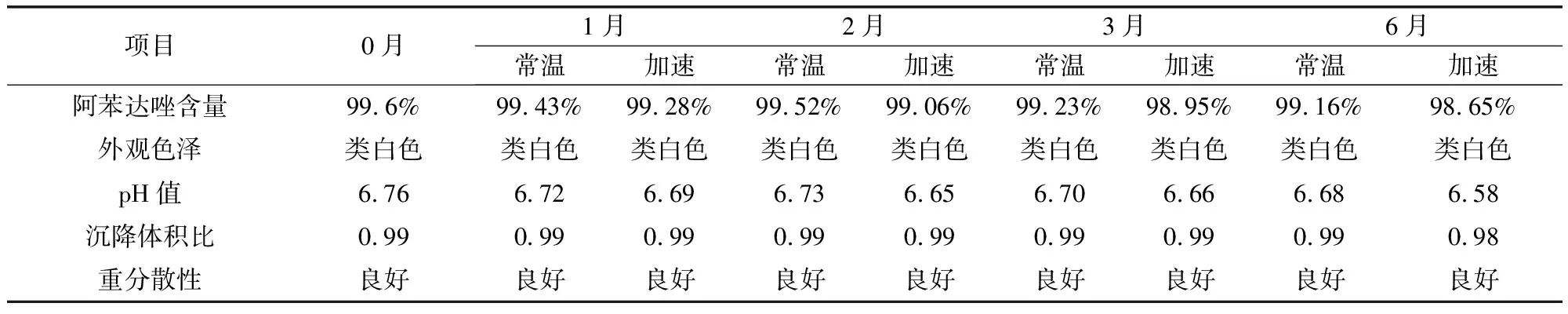
表4 阿苯達唑納米混懸液加速試驗結果
表5 阿苯達唑納米混懸液的物理穩定性
3.3 阿苯達唑納米混懸液在大鼠體內的動力學特征
3.3.1 方法專屬性 阿苯達唑亞砜ABZSX在本試驗所建立的色譜條件下,保留時間為9.7 min,基線平穩,藥物峰形良好,均能與雜質峰良好分離,檢測效率高。內標甲苯咪唑的保留時間為22 min,阿苯達唑保留時間為28 min。空白血漿色譜圖如圖2,空白血漿添加ABZSX(2ug/mL)、內標MBZ(10 μg/mL)、阿苯達唑ABZ(3 μg/mL)的色譜圖如圖3,樣品血漿色譜圖如圖4。
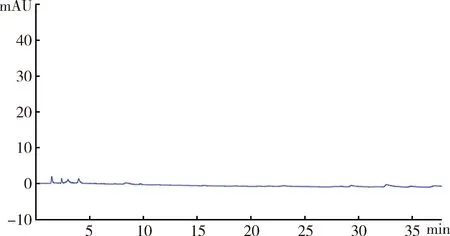
圖2 空白血漿色譜圖
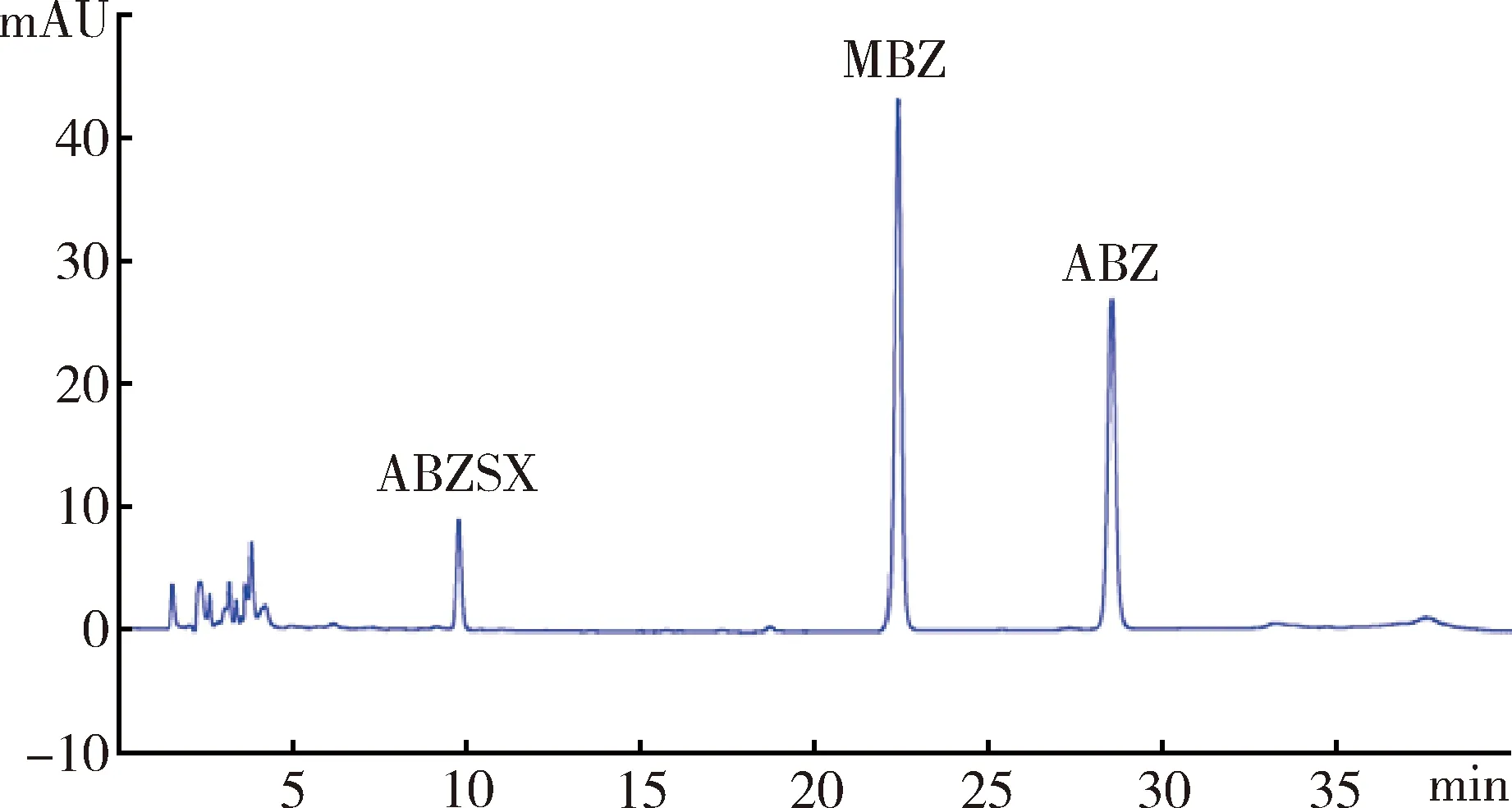
圖3 空白血漿中添加ABZSX、MBZ、ABZ色譜圖
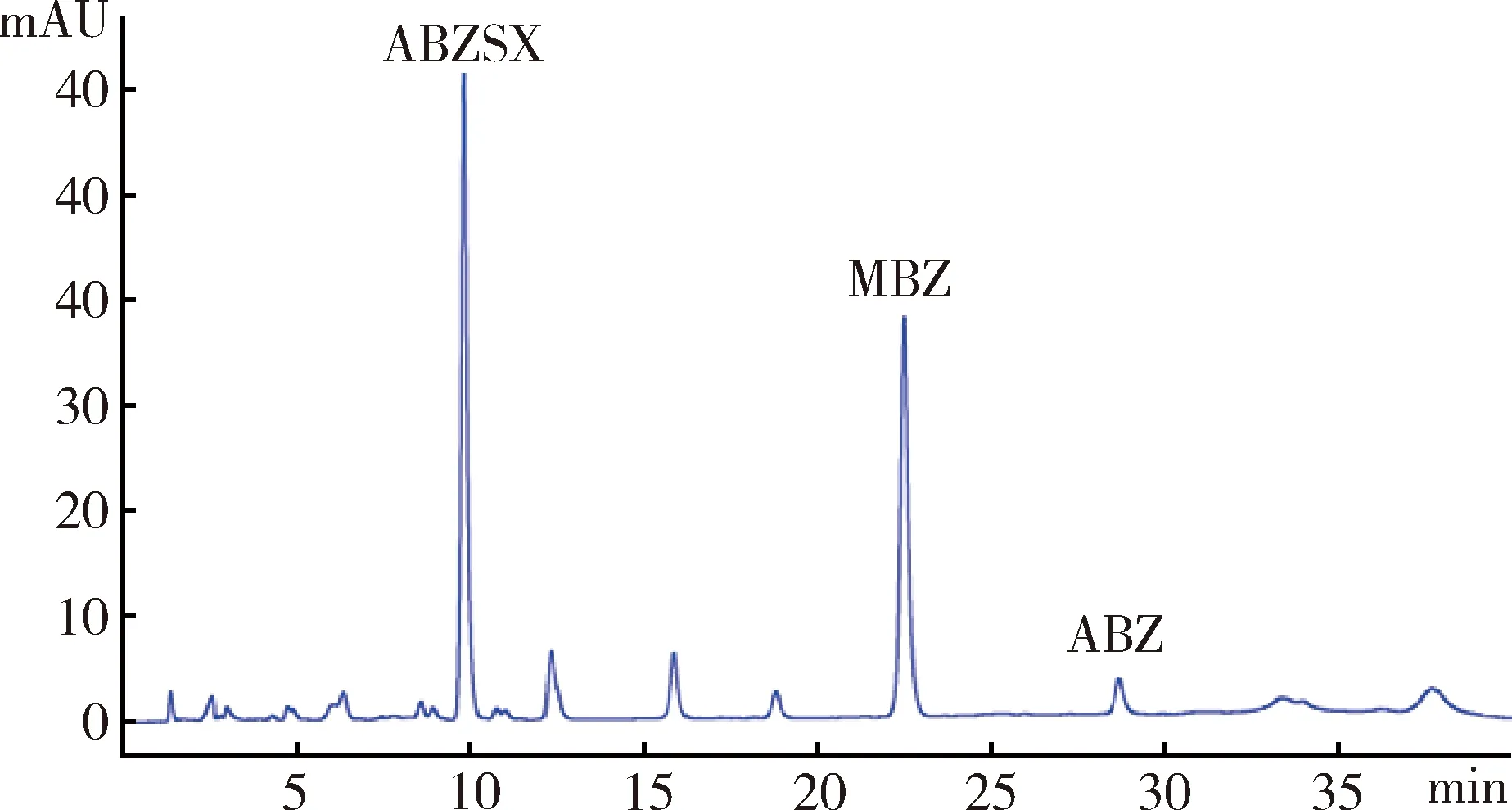
圖4 給藥后血漿中ABZSX、MBZ、ABZ色譜圖
3.3.2 血漿標準工作曲線 阿苯達唑亞砜(ABZSX)在 0.02~10 μg/mL濃度范圍內,血漿藥物濃度與ABZSX峰面積與內標峰面積二者比值呈良好的線性關系,回歸方程為y=0.2501x+0.0147,相關系數r為0.9992。
3.3.3 方法學驗證結果 阿苯達唑亞砜(ABZSX)在 0.02~10 μg/mL濃度范圍內,該分析方法的回收率為83.39%~95.41%;日內變異系數均在6.29%以內,日間變異系數均在6.19%以內,精密度良好;檢測限(LOD)、定量限(LOQ)分別為0.01、0.02 μg/mL。
3.3.4 藥物動力學數據 大鼠口服三種制劑的各時間點平均血藥濃度見表6,平均血藥濃度-時間曲線見圖5。采用非房室模型分析方法估算三種制劑的藥動學參數及相對生物利用度,具體藥物動力學參數見表7。
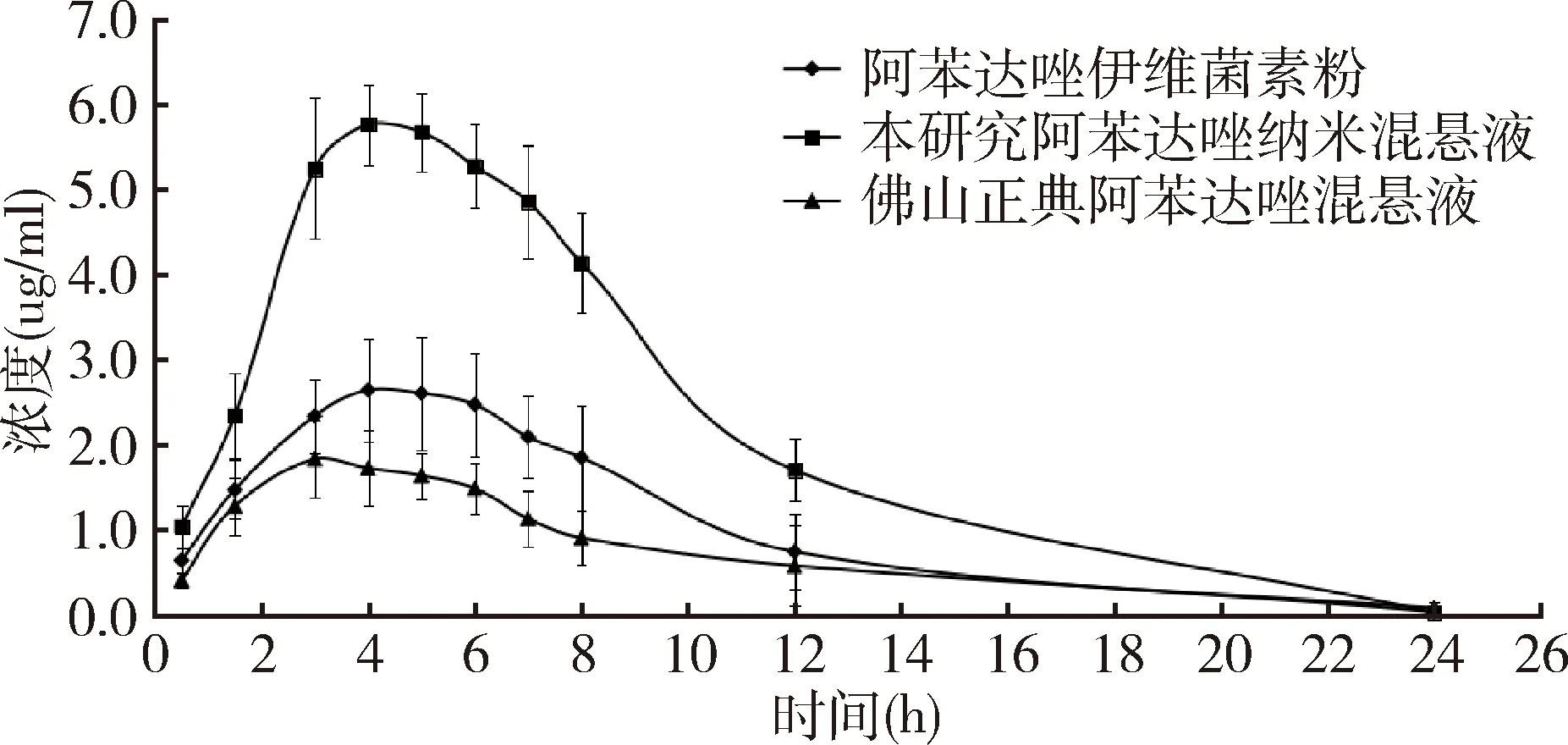
圖5 大鼠口服阿苯達唑伊維菌素粉及2種混懸液(45 mg/kg.bw)后ABZSX的血藥濃度-時間曲線

表6 大鼠口服阿苯達唑伊維菌素粉及2種混懸液(45 mg/kg.bw)后各時間點代謝產物ABZSX的血藥濃度(n=10)
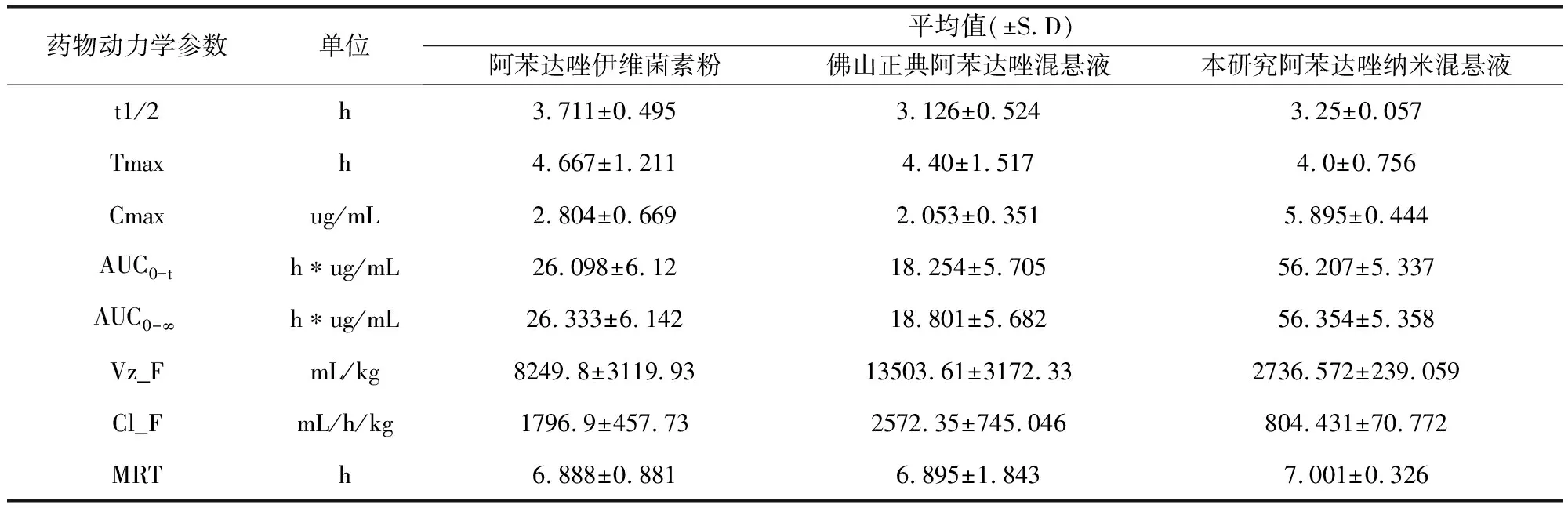
表7 大鼠口服阿苯達唑伊維菌素粉及2種混懸液(45 mg/kg.bw)后ABZSX的主要藥動學參數(n=10)
研制的阿苯達唑納米混懸液與阿苯達唑伊維菌素粉、佛山正典阿苯達唑混懸液的血藥峰濃度Cmax分別為5.895、2.804、2.053 μg/mL,藥物達峰時間 Tmax分別為 4.0、4.667和4.40 h,表明研制的阿苯達唑納米混懸液藥物峰濃度顯著高于阿苯達唑伊維菌素粉及佛山正典阿苯達唑混懸液,吸收速率明顯高于其他兩種制劑;且其對阿苯達唑伊維菌素粉、佛山正典阿苯達唑混懸液的相對生物利用度分別為214.0% 和299.74%,表明研制的阿苯達唑納米混懸液能顯著提高阿苯達唑的生物利用度。
4 討論與結論
納米晶體是提高難溶性藥物溶解度的有效策略之一[8],本研究納米混懸劑制備工藝主要是將“Bottom-up”技術中的沉淀技術與“Top-down”技術中的高壓均質技術聯合起來使用,即反溶劑法-高壓勻質法[9]。沉淀過程當中藥物在析出時會產生結晶態顆粒的納米晶體,或者產生無定型態的納米晶體,在此兩種形態的納米晶體基礎上,再次進行高壓均質會較容易獲得分散指數及粒徑更小的納米晶體。同時在藥物沉淀過程中通過高速分散器剪切,使析出的納米晶體通過初步剪切獲得較小粒徑,得到初混液,在將初混液進行高壓勻質,避免需要更多的均質化循環和更大的循環壓力使粒徑降低,可節約時間減低機器磨損。
納米混懸劑在配方上,改變了傳統沉淀法是將藥物溶解于有機溶劑中,穩定劑溶解至反溶劑(通常是水)中進行結晶析出的方法。傳統沉淀法的缺點是需要將藥物溶解在有機溶劑中,得到的納米混懸液會殘留有機溶劑,物理穩定性較差,易產生沉降聚集使藥物負載量低[10]。本研究未采用有機溶劑,而是將藥物溶解在助溶劑蘋果酸溶液中,在保證藥物性質穩定的加熱條件下,進行溶解后,加入穩定劑水溶液中進行結晶析出。該制劑中輔料蘋果酸可用于藥物制劑,有利于藥物在體內吸收、擴散,對機體有益。
本研究納米混懸劑的質量評價及制劑穩定性研究結果表明,其符合《中國獸藥典》2015版中對混懸液的質量要求,并在規定條件下進行加速試驗6個月,經檢測均符合質量要求,說明本研究阿苯達唑納米混懸液具有很高的物理穩定性。
阿苯達唑口服后,被代謝為阿苯達唑亞砜ABZSX、阿苯達唑砜ABZSN及ABZSO2-NH2,其中最主要代謝產物為ABZSX,它也是抗棘球蚴病的活性成分[11]。大鼠體內的藥物代謝動力學和相對生物利用度實驗結果表明,口服本研究的阿苯達唑納米混懸液與阿苯達唑伊維菌素粉、佛山正典阿苯達唑混懸液相比,體內的主要藥動學參數 Tmax、Cmax、AUC0-∞、Vz_F、Cl_F、t1/2均有顯著差異;本制劑的生物利用度顯著高于粉劑(相對生物利用度 F為214.0 %)、佛山正典產品(相對生物利用度 F為299.74 %)。阿苯達唑屬于濃度依賴性藥物,藥物濃度的提高將顯著提高其臨床治療效果。
本研究中,大鼠單劑量(45 mg/kg.bw)口服阿苯達唑納米混懸液后,納米混懸劑組Tmax為4.0 h,Cmax為5.895 μg/mL;粉劑組Tmax為4.667 h,Cmax為2.804 μg/mL;納米混懸劑相對粉劑組生物利用度為214.0%。與任潔如等[12]研究的大鼠單劑量(63 mg/kg.bw)口服阿苯達唑納米微粉的結果基本趨勢一致,即納米微粉組Tmax為3.15 h,Cmax為6.11 μg/mL;原料藥組Tmax為3.42 h,Cmax為3.20 μg/mL;納米微粉相對原料藥的生物利用度為157.0%;說明阿苯達唑納米制劑可提高藥物在體內的吸收速度和吸收量,以提高藥物生物利用度,增強藥物療效。
綜上,將阿苯達唑采用反溶劑法-高壓勻質法制備的納米混懸液,可獲得更小的藥物粒徑,與阿苯達唑伊維菌素粉、佛山正典阿苯達唑混懸液相比,藥物制劑粒徑的減小使其更易迅速均勻的分散于胃腸液中,在胃腸道內吸收迅速且吸收量增加,提高藥物生物利用度,增強驅蟲效果。本研究將對阿苯達唑的新劑型研發和臨床使用提供重要參考。
猜你喜歡
現代臨床醫學(2022年4期)2022-09-29 07:38:00
昆明醫科大學學報(2021年4期)2021-07-23 01:21:50
天津醫科大學學報(2019年6期)2019-08-13 07:04:34
云南醫藥(2019年3期)2019-07-25 07:25:14
現代檢驗醫學雜志(2016年5期)2016-08-20 03:16:56
海南醫學(2016年8期)2016-06-08 05:43:00
西南軍醫(2016年5期)2016-01-23 02:20:33
川北醫學院學報(2015年5期)2015-12-05 08:22:28
醫學研究雜志(2015年9期)2015-07-01 17:28:15
現代檢驗醫學雜志(2015年1期)2015-02-06 01:59:26
