構建硫酯官能團研究新進展
2022-07-06 03:12:44吳瑞鼎洪高健施湘君
浙江化工 2022年6期
吳瑞鼎,洪高健,施湘君
(浙江工業大學 長三角綠色制藥協同創新中心,浙江 杭州 310014)
硫酯類化合物廣泛存在于自然界,是有機合成研究中重要的酰基源以及結構轉換的中間體。膽固醇酯轉移蛋白(CETP)抑制劑達塞曲匹、生物素合成關鍵中間體、抗生素類藥物頭孢噻呋、乙酰輔酶A 及皮質類固醇替莫貝松等化合物中都能找到其身影[1](Scheme 1)。

Scheme 1
1 合成硫酯化合物的傳統方法
通過簡單高效的策略以獲得復雜的產品,是開發有機合成新手段的不竭動力。在過去幾十年中,人們進行了大量的努力,開發在合成各種具有生物應用前景的分子時獲得碳硫鍵的方法。由于其硫酯性質比酰鹵化合物穩定,在空氣氛圍中不易變質降解,因此在官能團轉化中扮演重要角色。由于巰基的易離去性,硫酯官能團也常作為酰基轉移試劑用于有機合成中。傳統構建硫酯鍵的方法是通過羧酸和醇在酸催化劑下加熱脫水縮合(Fischer 酯化法)、酰氯化合物與巰基親核加成[2]、酯交換或者通過DCC/EDC 等脫水劑在催化量的DMAP 存在條件下[3](Steglich 酯化法),進行縮合得到想要的官能團片段(Scheme 2)。但考慮到制備酰氯的過程中會用到草酰氯或者氯化亞砜等危險試劑,且酰氯中間體易吸濕、性質不穩定,酰化試劑對生產設備具有腐蝕性,存在潛在風險,運用中仍存在諸多不便。從原料易得、反應高效、處理簡單等角度考慮,開發新的構建硫酯官能團的方法迫在眉睫。本文介紹最近幾年合成硫酯化合物的新方法和新途徑。
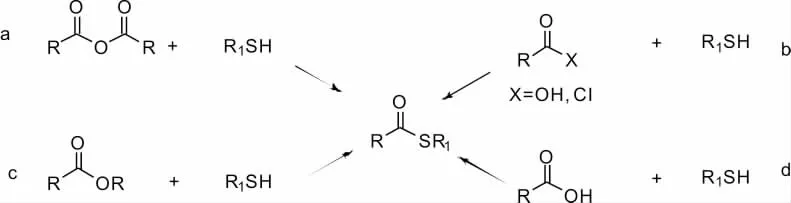
Scheme 2
2 構建硫酯鍵的研究新進展
2.1 BrΦnsted 酸介導的偶聯反應
羧酸與硫醇反應中的平衡不利于硫酯的形成,Iimura 等[4]通過篩選發現10 mol%的TfOH 在甲苯中共沸回流條件下能夠催化分子間的交叉酯化偶聯反應(Scheme 3),原子經濟性好且能顯著降低反應的活化屏障。反應不僅適用于一級和具有空間位阻的二級脂肪族底物,而且針對芳香族硫醇,以優異的產率生成相應的硫酯。但當采用TiCl4、ZrCl4、SnCl4等Lewis 酸催化時則難以實現反應,這表明過程中需要BrΦnsted 酸的質子參與。

Scheme 3
El-Azab 等[5]的研究發現,在CF3COOH 的催化下苯甲酸和苯硫酚能夠形成碳硫鍵(Scheme 4)。該反應條件溫和,通過篩選,在60 ℃下使用CH3CN 作溶劑最佳,反應收率達到95%以上。通過形成酸酐型原位中間體,巰基進攻并脫去三氟乙酸進一步轉化為相應的硫酯。該反應還成功拓展至羥基醇,得到90%以上的產率。局限性在于該反應羧酸底物限定于芳基性化合物,對于脂肪族化合物產率較低。
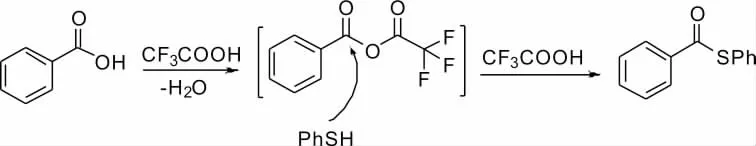
Scheme 4
2.2 銅催化烷基苯化合物與硫酚脫氫偶聯反應
Ali 等[6]報道了苯硫酚與烷基苯化物之間偶聯生成硫酯類化合物的新方法。該方法使用二水合醋酸銅作為催化劑,氧化劑優選為過氧化叔丁醇(Scheme 5),通過將烷基苯化合物氧化成苯甲酰自由基,成功實現烷基苯化合物與硫酚化合物的脫氫偶聯。該方法的優勢在于底物適應性好,不需要定位基團即可實現反應的高選擇性。但反應收率偏低,最優條件下僅為73%,且氧化劑的使用量較大。

Scheme 5
2.3 銅催化芳酰肼與二硫化物的氧化硫代酯化反應
Xie 等[7]通過系統研究,在氰化亞銅的存在下,芳酰肼與二硫化物能被氧化劑偶聯生成相應的硫酯(Scheme 6)。該方法穩定,以容易獲得的芳酰肼作酰基源,相對無味的二硫化物作硫源,避免了巰基化合物的惡臭氣味,有研究開發價值。但優選使用的催化劑氰化亞銅毒性大,存在較大安全隱患。若對于產率沒有較高要求時可考慮使用氯化亞銅替代。

Scheme 6
2.4 銅催化α-羰基甲酸與二硫化物的脫羧偶聯酯化反應
Rong 等[8]報道了在溫和的條件下Cu 催化α-羰基甲酸與二苯二硫醚脫羧偶聯成酯的方法(Scheme 7)。此方法如果沒有催化劑或氧化劑,反應是難以發生的。其中CuO 表現出最好的催化效率,以過硫酸銨為氧化劑,DMSO 與H2O(體積比為5:1)的混合溶劑為最佳,提供了一種簡便、產率較高的合成硫酯化合物的方式。

Scheme 7
此反應可能的機理是α 羰基甲酸在銅(Ⅱ)催化劑存在下生成甲酰自由基,然后自由基與二硫醚進一步反應脫硫醇或硫酚得到硫酯。銅(Ⅰ)離子接下來將被過硫酸銨氧化為銅(Ⅱ),并返回到反應中(Scheme 8)。

Scheme 8
2.5 銅催化芳醛與硫醇之間的硫酯化反應
Yi 等[9]報道了在水相中,過氧化叔丁醇(TBHP)作氧化劑的條件下,Cu 催化芳醛和硫醇之間的C-S 鍵形成(Scheme 9)。銅離子和亞銅離子等都能催化此反應,其中以氯化亞銅的效率最高。底物適應性廣,包括氯、三氟甲基、溴、碘、腈、酯和噻吩在內的官能團都可以在所采用的反應條件下進行反應。芳醛與芳硫醇和烷基硫醇都能偶聯,以中等至良好的產率提供相應的硫酯。該反應在水中進行,反應條件綠色,對環境友好。

Scheme 9
2.6 偕二氟烯烴自催化參與的硫羰基化反應
Jiang 等[10]報道了由偕二氟烯烴參與的硫羰基化反應(Scheme 10)。由于氟原子對烯烴α-碳的σ-吸電子誘導效應,芳基硫醇與偕二氟烯烴的區域選擇性親核加成反應在沒有任何催化劑的情況下提供線性α,α-二氟烷基硫醚。隨后的轉化反應不同于典型的C-F 鍵活化通常依賴于Si-F、B-F、P-F、和金屬-F 鍵等以克服C-F 鍵的高解離能,兩個C-F 鍵在無金屬和無添加劑條件下斷裂。這是迄今為止偕二氟烯烴的首次硫羰基化反應。該反應底物適應性強,芳環上無論是吸電子還是供電子基團都具有良好的耐受性。反應機理可能是偕二氟烯烴與硫酚生成物α,α-二氟烷基硫醚,然后被微量偕二氟烯烴水解生成的氫氟酸自活化,促進反應的進行。
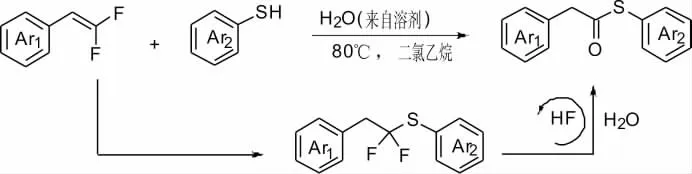
Scheme 10
2.7 TBAI/K2S2O8 促進的烷基酮與芳香族硫醇/二硫化物的氧化硫酯化反應
α-酮硫酯是重要的C-S 鍵衍生物,也是有機合成中進一步轉化非常有用的試劑。Hu 等[11]報道了由四丁基碘化銨/過硫酸鉀(TBAI/K2S2O8)促進的甲基酮與芳香族硫醇/二硫化物的氫鍵氧化硫酯化反應(Scheme 11)。該反應為芳香族硫醇官能化合成α-酮硫酯提供了一種簡單有效的方法。

Scheme 11
2.8 DBH 和二甲基亞砜介導的烯烴氧化硫代酯化反應
Hua 等[12]選擇苯乙烯作為模板反應并篩選了反應條件(Scheme 12)。首先,將苯乙烯在室溫下用DBH 和Na2CO3在無水二甲基亞砜中處理10 h,產物收率為48%。為了提高產物的收率,篩選了幾種堿性條件。結果表明,用NaHCO3提供堿性條件是穩定的,并在40 ℃下獲得最佳產率。強吸電子基團(NO2)取代的苯乙烯未能生成所需的產物,這可能是電子云密度降低所致。該反應對于稠環、雜環、脂肪族底物適應性良好,為烯烴的硫酯化提供了新途徑。

Scheme 12
2.9 亞胺介導的氧化硫代酯化反應
Mupparapu 等[13]報道了一種高效合成α-酮硫酯的方法。該反應的特點是吡咯烷能夠通過形成亞胺中間體介導α-羰基醛和硫醇之間的反應(Scheme 13),以中上的產率得到所需的產物。該反應生成硫酯鍵中的氧經控制實驗,證實來自于空氣中的氧氣,不需要外部氧化劑或金屬催化劑。
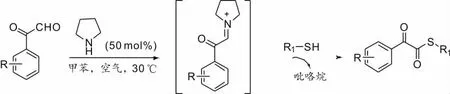
Scheme 13
2.10 鎳催化芳基碘化物的硫酯化反應
Iranpoor 等[14]報道了以六羰基鉻作為氧源,由Ni 催化芳基碘化物的硫代酯化反應(Scheme 14)。該反應由NiCl2作催化劑,K2CO3提供堿性條件,在無需配體的情況下高效實現硫酯化反應,反應生成部分硫醚副產物(10%)。該反應同樣適用于醇類、胺類底物,可開發性較好。
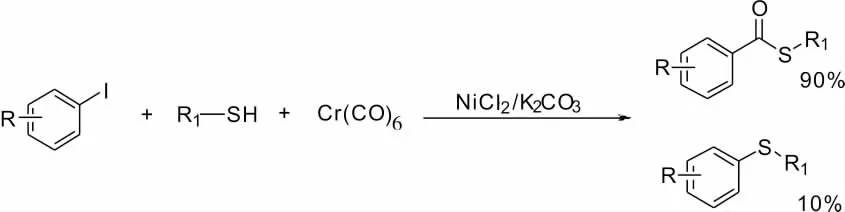
Scheme 14
2.11 硫代硫酸鈉五水合物作為硫源的硫酯化反應
Liao 等[15]報道了由Na2S2O3·5H2O 作為硫源,一鍋法實現有機鹵化物與芳基酸酐合成硫酯化合物的方法(Scheme 15)。該反應溶劑優選為DMF,條件溫和且無需金屬催化,底物適應性強。

Scheme 15
在此反應中,芳基酸酐首先與硫代硫酸鈉進行親核酰基取代反應,生成相應的Bunte 鹽作為中間體。向反應混合物中加入有機鹵化物后,鹽自發形成硫芳酰陰離子。最后,硫芳酰化物與有機鹵化物反應得到相應的硫酯產物(Scheme 16)。

Scheme 16
3 總結和展望
硫酯基作為一種十分重要的官能團,是天然活性成分的分子骨架,在醫藥、農藥等化學品中具有關鍵作用。酸和硫醇縮合的酯化反應是傳統合成硫酯的方式,目前的合成方法有些具有底物適應性差、能耗高、產率低等問題。因此不斷探索廉價高效的反應體系對于硫酯化合物的合成至關重要。目前已探索出以芳酰肼、芳醛、α-羰基甲酸、α-羰基醛、烷基酮等為酰基底物,硫醇、二硫化物、二甲基亞砜、硫代硫酸鈉五水合物等為硫源的方法。但其中有些用到了金屬離子催化和一些危害較大的氧化劑,收率仍有提升的空間。相信隨著相關研究工作的深入,越來越多的方法將不斷涌現。
