富鎳正極材料LiNi0.8Co0.1Mn0.1O2 的制備和改性研究進展
2022-07-06 03:12:44田崇旺徐立鵬包春江冷旭寧
浙江化工 2022年6期
田崇旺,徐立鵬,包春江,冷旭寧
(1.聊城大學 機械與汽車工程學院,山東 聊城 252000;2.山東黃海科技創新研究院,山東 日照 276800)
當今世界面臨著兩大能源挑戰,一是將傳統能源(煤炭、天然氣等)發電轉向可持續能源(風能、太陽能等)發電,二是將燃油汽車轉向純電動汽車[1]。鋰離子電池技術正是解決這兩大問題的關鍵技術之一。在三元鋰離子電池中,正極材料LiNi0.33Co0.33Mn0.33O2和LiNi0.5Co0.2Mn0.3O2雖然具有較好的穩定性但能量密度相對較低。眾所周知,富鎳三元正極材料隨著鎳含量的提升,其比容量逐漸增大。LiNi0.8Co0.1Mn0.1O2(簡稱NCM811)正極材料與已商業化的LiNi0.6Co0.2Mn0.2O2相比,雖然其熱穩定性及容量保持率不理想,但憑其超高的比容量、低廉的成本,是目前滿足我國新能源戰略規劃,實現“中國制造2025”動力電池發展規劃的理想材料[2]。LiNi0.8Co0.1Mn0.1O2已經成為高容量新型鋰離子正極材料的研究重點之一[3]。
1 三元正極材料LiNi0.8Co0.1Mn0.1O2 的制備方法
目前,合成LiNi0.8Co0.1Mn0.1O2正極材料的方法有共沉淀法、高溫固相法、水熱法、溶膠-凝膠法等。盡管制備鋰離子電池正極材料的方法有許多種,但不同制備工藝對材料的粒徑、相結構和比表面積影響較大,進而造成其電化學性能差異明顯。
1.1 共沉淀法
共沉淀法是目前工業合成三元材料前驅體的主要方法。先將原料按比例配制成混合金屬溶液,然后使用絡合劑和沉淀劑將溶液中的金屬離子共沉淀出來。通過控制反應條件,制備的前驅體形貌規整且粒度可控,前驅體的形態對正極材料的電化學性能至關重要[4]。
Zheng 等[5]采用霧化共沉淀法成功合成LiNi0.8Co0.1Mn0.1O2。經研究發現:其在高倍率5 C(1 C=200 mA·g-1)下放電容量高達157.38 mAh·g-1;經2 C,100 次循環后,容量保持率仍高達88.5%。分析認為,霧化共沉淀法減小了顆粒尺寸,縮短了Li+的擴散路徑,增加了與電解液的接觸面積。
Savina 等[6]采用不同過渡金屬鹽和氫氧根共沉淀法制備了(1-x)LiNi0.8Co0.1Mn0.1O2·xLi2SO4(x=0.002~0.005)復合正極材料。在硫酸鹽、富氧條件下制備的正極材料性能最佳。分析認為非晶態Li2SO4提高了NCM811 初級粒子界面斷裂韌性,抑制了高電壓相變和長時間循環后積累的反位無序,改善了二次團聚體的力學性能。
Zhou 等[7]利用綠色復合螯合劑檸檬酸和谷氨酸鈉制備了Ni0.8Co0.1Mn0.1(OH)2前驅體。研究發現:該前驅體經H2O2溶液預氧化,并在無CO2的流動空氣氣氛下,于780 ℃下燒結20 h 制備的正極材料性能最優。
1.2 水熱與溶劑熱合法
水熱法是濕化學法合成的一種,在高溫高壓條件下利用溶液中物質的化學反應進行合成的方法。水熱法具有反應易于控制、產品結晶度高、反應溫度較低等優點[8]。
Zeng 等[9]采用草酸鹽水熱法合成了LiNi0.8-Mn0.1Co0.1O2,研究發現,在100 ℃下20 min 內Ni2+、Mn2+和Co2+可快速有效沉淀,極大地縮短了制備時間。同時在1 C 時,初始放電容量為183.0 mAh·g-1,初始放電庫倫效率為80.29%。在6 C、10 C 和15 C 分別獲得了約138 mAh·g-1、120 mAh·g-1和95 mAh·g-1的可逆容量。
Hendri 等[10]以硫酸鹽為原料,采用水熱法制備出LiNi0.8Mn0.1Co0.1O2,研究發現,隨著溫度的升高,晶粒尺寸逐漸增大,得到的晶粒尺寸在45.37~46.74 nm 之間。水熱溫度為190 ℃時,正極材料的晶粒尺寸達到最大,在0.05 C 時,正極材料的比容量最高達216 mAh·g-1。
在富鎳三元正極材料制備的眾多方法中,共沉淀法是最成熟的方法之一,該方法可以很好地控制材料的形貌、粒徑和比表面積等結構參數,同時可以保證3 種元素達到原子級水平的均勻混合。在共沉淀制備方法中,以硫酸鹽為過渡金屬鹽時,硫以Li2SO4的形式存在于晶界和晶間接觸處,顯著增強了容量保持力,提高了正極材料的倍率性能和初始放電容量。碳酸鹽共沉淀工藝雖然成本較低,但穩定性較差,產物粒徑不均勻。此外,碳酸鹽前驅體振實密度比氫氧化物前驅體低,而較低的振實密度也嚴重限制了正極材料能量密度的發揮。而氫氧根共沉淀的溶度積(Ksp)較碳酸鹽共沉淀的小,氫氧根共沉淀反應進行得更完全,更容易控制前驅體的形貌。在結鋰燒結時,采用碳酸鋰和富氧氣氛下經過低溫預處理和高溫燒結工藝合成的LiNi0.8Co0.1Mn0.1O2最優。
2 三元正極材料的改性方法
雖然三元正極材料的比容量隨著鎳含量的增加逐漸增加,但鈷和錳含量的降低不可避免地導致許多問題。例如:(1)Ni 含量的增加會增加正極材料的陽離子混排程度,降低首次庫倫效率,同時在反應中需要加入過量的鋰鹽,影響加工性能;(2)Co 含量的降低使材料的導電性和循環倍率降低,影響電池的使用壽命;(3)Mn 含量的降低直接影響材料的穩定性;(4)正極材料容易與電解液發生反應,腐蝕正極材料,降低電池的循環性能[11-12]。因此,利用離子摻雜、表面包覆、共混改性和梯度設計等方法對三元正極材料進行改性以提高材料性能非常重要。
2.1 離子摻雜
離子摻雜是一種常見的改性方法,摻雜可提高材料的晶格能以穩定材料的晶體結構,合適的離子摻雜有助于減輕材料的陽離子混排程度,抑制Janh-Teller 效應,維持材料的結構穩定性,進而有效提高材料的電化學性能[13]。離子摻雜主要有3 種方式:陽離子摻雜、陰離子摻雜和多元素共摻雜。
Kim 等[14]通過固相反應制備摻雜F-的正極材料。研究發現,F-在氧位的取代可使過渡金屬與F之間形成強鍵,增強結構穩定性,抑制相變。此外,F-取代氧導致部分過渡金屬的還原,增加了其離子半徑,提高了鋰離子的擴散系數,從而在高電流密度下具有優越的循環性能。
Chu 等[15]采用固相合成法在鋰位和過渡金屬位上同時摻雜Nb。結果表明,在1%的摻雜量時性能最優,在2 C、電壓范圍為2.7~4.5 V、初始放電容量為202.8 mAh·g-1,200 次循環后放電保留率仍為81%,明顯優于原始正極材料。分析認為,Nb 摻雜一方面導致過渡金屬層中離子的平均半徑增大,另一方面由于需要更多的Ni2+來平衡Nb的高價位,實現電中性的約束,導致Ni2+含量增加。在富Ni 層狀陰極中,實現雙位點摻雜不僅可以抑制Li/Ni 無序化,促進鋰離子輸運,而且可以穩定層狀結構,防止結構局部塌陷和結構變形。
Tao 等[16]采用共沉淀法制備了Mg 摻雜的正極材料。研究發現,正向濃度梯度摻雜具有更優的電化學性能,在5 C、電壓范圍為2.7~4.5 V 下,正向濃度梯度摻雜的正極材料放電容量為160.3 mAh·g-1,優于其他摻雜方式。摻雜量為3%時,在0.1 C、電壓范圍為2.7~4.5V 下,放電容量為240.1 mAh·g-1,遠高于其他正極材料。研究表明,Mg2+的加入增大了層間距,使Li+的嵌入和脫出更加容易,在充電過程中,Ni2+被氧化為Ni3+/Ni4+,導致層狀結構局部坍塌,而Mg2+占據了Li+的位置,起到了支柱作用,提高了循環性能。
Xu 等[17]采用熔鹽輔助生長法成功合成K+摻雜的LiNi0.8Co0.1Mn0.1O2正極材料,鉀離子的摻雜可有效降低Li+/Ni2+陽離子的混排并提高層狀晶體結構的有序性,同時有助于晶體中Ni3+和Co3+的形成。此外,在100 次循環后,正極上P-、F-和Ni3+的覆蓋量均低于原始材料。結果表明,鉀離子的摻雜能有效抑制充放電循環過程中的副反應、金屬陽離子的溶解和電解質的分解。
2.2 表面包覆
表面包覆是指在正極材料表面均勻地包覆一層致密的惰性材料,并在正極材料和電解液之間形成物理屏障,避免正極材料與電解液直接接觸,有效抑制副反應的發生。包覆材料可分為氧化物、氟化物、磷酸鹽、鋰化物等。氧化物包覆材料最為普遍,可以顯著提高循環性能和熱穩定性,而氟化物包覆材料可以抵抗HF 腐蝕,避免正極材料的破壞,同時包覆層還可以有效緩解結構的惡化[18]。
Liu 等[19]通過溶膠-凝膠法和溶劑熱法制備出SnO2和Li2SnO3包覆的LiNi0.8Mn0.1Co0.1O2正極材料。研究認為,SnO2和Li2SnO3涂層均能有效抑制正極/電解質界面的副反應,降低界面電阻,增強鋰離子在正極/電解質界面的遷移。
Kimura 等[20]成功制備了Al2O3涂層的NCM811正極材料,并與粉末Al2O3和NCM811 混合的正極材料進行對比。研究表明,Al2O3和NCM811 混合不存在協同作用,只有機械化學處理使Al2O3和正極材料顆粒之間緊密接觸才是提高正極材料循環性能的關鍵。同時鋁固溶層抑制了沉積層的生長,HF 被Al2O3捕獲形成AlF3,抑制了類NiO層的生長,從而提高了正極材料的循環性能。
Peng 等[21]采用聚丙烯酸(PAA)對正極材料進行表面改性,研究表明,包覆后的正極材料不僅可提高材料的循環性能,還可以延長儲存性能。在極端環境儲存6 d 后,改性后的正極材料的初始能量密度為115.8 mAh·g-1,較原始材料的53.3 mAh·g-1提高了近兩倍。分析認為,穩定的PAA涂層使NCM811 材料對潮濕空氣的敏感性降低,相同存儲過程后的改性材料中LiOH 和Li2CO3雜質較少,大大提高了材料的存儲性能。
2.3 核殼結構及濃度梯度
核殼結構及濃度梯度是近年來研究的熱點。核殼結構一般指用具有電化學性能較好的材料包覆在富鎳材料的表面,以提高正極材料的穩定性。研究發現核殼并不能較好地結合,在多次循環后容易出現裂紋,嚴重影響電化學性能。濃度梯度是指正極材料的晶體顆粒Ni 含量由內到外逐漸減少,而Mn 含量由內到外逐漸增多。表面Mn 離子含量的增多可以提高材料的穩定性,核殼結構及濃度梯度制備的材料性能較優,但工藝過程較復雜,效率較低。
Wu 等[22]利用C4H8N2O2制備出核殼結構的Li Ni0.8Co0.1Mn0.1O2,材料芯部鎳含量高,表面錳含量高。研究發現,C4H8N2O2與Ni 的配比為0.03:1 時制備的正極材料,在5 C、電壓范圍為3.0~4.5 V下,500 次循環后,放電容量保持在100 mAh·g-1,遠高于原始LiNi0.8Co0.1Mn0.1O2正極材料的60 mAh·g-1,同時核殼結構在高倍率和高溫條件下也具有較好的電化學性能。
Shin 等[23]采用共沉淀法合成核-多殼結構正極材料。研究發現,與傳統的均質陰極材料相比,增加殼的數量可以提高容量保持和倍率能力。殼層的高穩定性通過降低與電解液的反應來改善電化學性能。微壓痕研究表明,顆粒破碎力與陰極材料的一次顆粒組成和二次顆粒大小有關。斷裂力越高,在陰極電極制備的壓延過程中,顆粒越不易開裂,從而抑制了表面積的增加。
Lin 等[24]采用共沉淀法合成了鎳元素濃度梯度的Ni0.8Mn0.1Co0.1(OH)2前驅體,并進一步利用較強的Mn-O 鍵,控制燒結氣氛合成Ni 價態濃度梯度的正極材料。改性后的正極材料,在0.2 C、電壓范圍為2.7V~4.4 V,經過100 次循環后,其比充放電容量在175 mAh·g-1以上,遠高于傳統正極材料的142 mAh·g-1,正極材料的容量保持能力得到了極大改善,見圖1。研究結果表明,建立一個氧化態梯度,將電容性較高但穩定性較差的Ni3+隱藏在二次粒子表面,是優化高鎳含量陰極材料的可行方法。
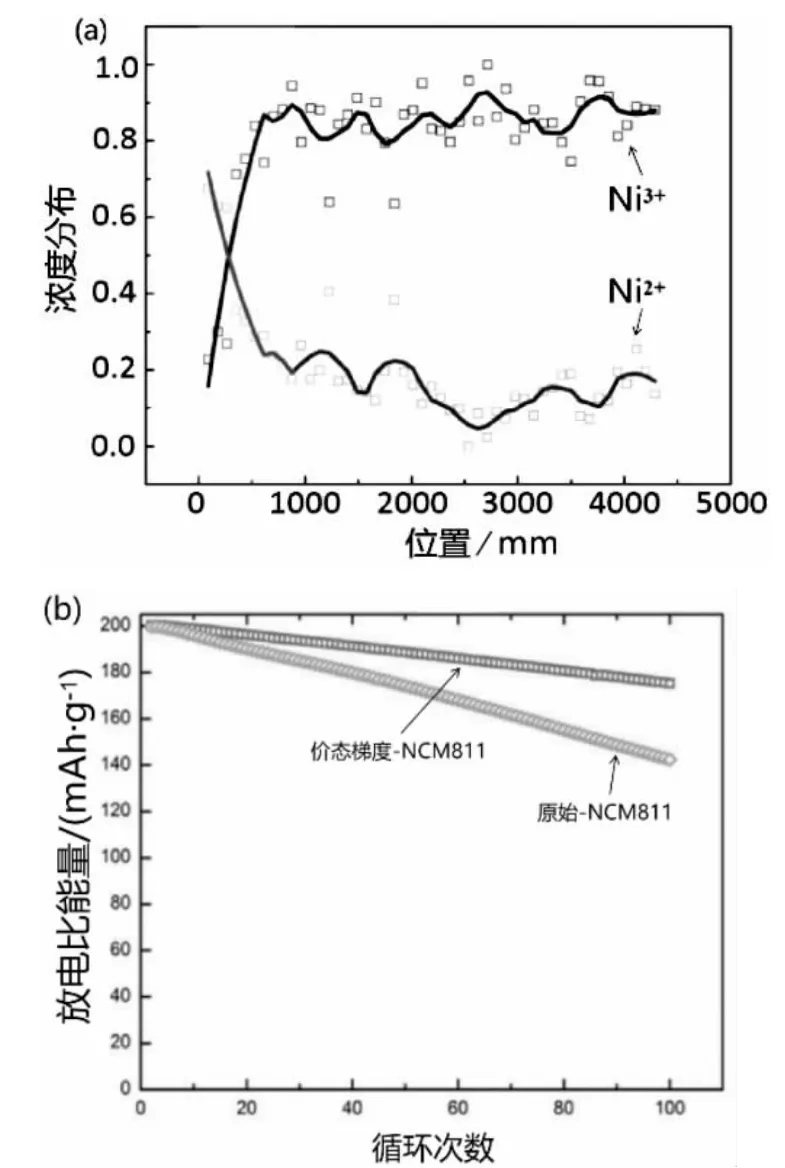
圖1 核-殼結構Ni 價態含量(a)及循環性能(b)[24]
3 結語
隨著市場對電池能量密度需求的不斷提高,三元電池的市場化逐步向高鎳型電芯轉變,然而鎳含量的不斷增加導致鋰電池循環壽命、安全性能不斷降低,嚴重制約著富鎳鋰電池的商業發展。
為解決上述問題,必須對三元正極材料進行改性設計。改性設計可采用:(1)單晶化設計,單晶化后的材料比表面積小、振實密度高、循環性能和熱穩定性較好;(2)協同改性,如Al2O3包覆和Zr 摻雜協同改性;(3)改善改性工藝,如采用原子層共沉淀包覆、石墨烯摻雜等;(4)采用核殼結構或濃度梯度設計,使正極材料同時擁有高容量和高安全性;(5)提高截止電壓,進一步提高電池能量密度,如W 元素的摻雜等。
雖然富鎳三元正極材料LiNi0.8Mn0.1Co0.1O2在安全性和循環壽命方面還存在不足,但隨著制備工藝的不斷優化,改性手段的不斷提升,企業與國家巨額資金的不斷投入,富鎳正極材料的潛能不斷被挖掘,安全性能將不斷提高,制造成本也必然不斷下降。富鎳三元正極材料有望在新能源、通信、軍事、儲能設備等領域得到廣泛應用,占據更多的市場份額。
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
中國塑料(2016年12期)2016-06-15 20:30:07
中國塑料(2016年5期)2016-04-16 05:25:36
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
中國塑料(2015年4期)2015-10-14 01:09:19
