早發性癲癇腦病患兒SCN1A CDKL5基因突變特點與臨床特征
2022-07-04 11:02:42廖趙妹蔡思銘陳劍標呂華龍雷智賢
河北醫學 2022年6期
廖趙妹, 蔡思銘, 陳劍標, 呂華龍, 雷智賢
(1.海南醫學院第二附屬醫院兒科一區, 海南 海口 5701002.海南醫學院附屬兒童醫院兒科, 海南 海口 570100)
癲癇(epilepsy)是一種受到多種因素影響造成的腦內異常過度神經元活動所引發的短暫性放電癥狀疾病,由于異常放電部位不同,擴散網絡堵塞息息相關,因而患者疾病發作時的表現形態、感覺知覺、精神狀態和行為意識均有不同[1,2]。早發性癲癇腦病(EOEE)是在患兒出生后幾個月內出現的癲癇疾病,由于患兒腦部及機體功能發育尚不完全,疾病發作時的頻繁放電異常將導致腦部發育遲緩或感知覺、認知功能、運動功能障礙。癲癇類疾病大多存在多種病因,在目前已知的癲癇類疾病中,大田原綜合征、早期肌陣攣腦病、嬰兒痙攣癥、Dravet綜合征以及各類尚未命名的非特異性癲癇腦病,其發病機制除了特定狀態下出現的遺傳、腦損傷、感染等因素,其他較為復雜的臨床癥狀原因尚且不明[3]。基于此,本實驗選取我院2018年4月至2019年4月期間收治的40例早發性癲癇腦病患兒臨床資料作為研究對象,采用基因篩選技術檢查EOEE患兒出現SCN1A、CDKL5基因突變的臨床特征,旨在為EOEE患兒臨床診療提供參考依據。具體研究報告如下:
1 資料與方法
1.1一般資料:選取我院2018年4月至2019年4月期間收治的40例早發性癲癇腦病患兒臨床資料作為研究對象,采集患兒父母外周血進行sanger測序排查。納入標準:①患兒出生1年半內出現驚厥現象;②可合并有多類型癲癇發作情況,在常規癲癇疾病藥物作用下疾病未好轉;③表現為腦智力落后或運動功能停滯等。排除標準:①遺傳性代謝障礙;②圍生期內發生的腦部損傷;③孕期內出現宮內感染;④經腦部影像學診斷為腦部結構異常所引發的新生兒癲癇。本次實驗已征得本院醫學倫理會批準同意,所有患兒家屬均對本實驗目的及相關內容知情,并自愿簽署知情同意書。
1.2方法:在家屬積極配合下采集患兒以及父母外周血各3mL,采集完成后用二胺四乙酸(EDTA)抗凝試管保存送往基因檢測中心進行癲癇基因包檢測。患兒外周血采用靶向二代測序技術分析患兒癲癇基因,父母外周血采用sanger測序排查,根據現有人類基因突變數據庫等數據對比分析,確定患兒存在致病可以和疑似致病可能的基因突變。此外,檢測為基因突變陰性的患兒采用多重連接探針擴增技術(MLPA)行大片基因段變異檢查,以此排除單核苷酸多態性存在。采用TIANGEN DNA提取試劑盒(北京天根生化科技有限公司)檢測患兒外周血所提取的全基因組DNA,具體步驟按照試劑盒說明書嚴格執行。獲取截至本實驗時間的最新文獻資料和數據庫資料,將本實驗中靶向二代測序技術分析所得的人類癲癇相關基因,轉入基因測序包(京德易東方轉化醫學研究中心有限公司),對所有癲癇相關基因進行文庫整理和編碼測序[4,5]。
1.3臨床表現:對所有癲癇陽性基因突變患兒的臨床表型資料進行分析,包括患兒的首次癲癇發作時間、癲癇發作類型、用藥情況、智力發育和運動情況等。
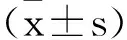
2 結 果
2.1一般資料:本研究中,患兒男12例,女28例,疾病發作并接受診療時間為出生6d至18個月(10.11±4.29)個月。經診斷,出現嬰兒痙攣癥患兒23例,非特異性EOEE患兒10例,Dravet綜合征4例,大田原綜合征患兒3例。所有患兒父母均排除近親婚配生育,且家族內未發現癲癇或神經疾病史。
2.2SCNIA、CDKL5基因突變特點:本研究發現32例(80.00%)SCNIA、CDKL5基因突變,有17例(42.50%)患兒SCNIA基因突變,15例(37.50%)患兒CDKL5基因突變,其中男11例(SCNIA基因突變5例,CDKL5基因突變6例),女21例(SCNIA突變12例,CDKL5突變9例)。采用Sanger測序法檢測基因突變的類型,發現17例(42.50%)錯義突變,3例(7.50%)插入突變,3例(7.50%)無義突變,3例(7.50%)剪切位點突變,6例(15.00%)微小缺失突變,未發現大片基因段缺失,見表1。所有患兒父母外周血基因檢測均未發現SCNIA、CDKL5基因突變,表明患兒SCNIA、CDKL5基因為新生突變。

表1 SCNIA CDKL5基因突變特點n(%)
2.3SCNIA、CDKL5基因突變患兒臨床特征:32例SCNIA、CDKL5基因突變的患兒中,患兒均在10d至3個月內第一次發病,且患兒發病時可出現兩種以上癲癇發作類型。有27例(84.38%)患兒首次發病出現無熱驚厥,6例(18.75%)患兒伴有強直陣攣,3例(9.38%)患兒伴有全身痙攣,隨著病情發展有5例(15.63%)患兒出現痙攣。在藥物方面,根據患兒具體情況對患兒施以2~8種抗癲癇類藥物治療,其中疾病發作低于50%并被認定為有效藥物治療的有:托吡酯27例(67.50%),丙戊酸鈉19例(47.50%),氯硝西泮17例(42.50%)。此外,有2例(5.00%)患兒經生酮食療,但臨床療效不明顯。32例SCNIA、CDKL5基因突變患者存在不同程度的智力發育落后情況,尚無患兒具備有意義語言和獨立行走能力,21例(65.63%)患兒獨坐時間明顯低于同年齡正常兒童,28例(87.50%)患兒具有孤獨癥征兆,11例(34.38%)存在手足功能低和身體發育緩慢現象。見表2。

表2 SCNIA CDKL5基因突變患兒臨床特征
3 討 論
早發性癲癇腦病(EOEE)是一類癲癇疾病的綜合,除了非特異性癲癇腦病,目前已確定的有Dravet綜合征、嬰兒痙攣癥、大田原綜合征和陣攣性腦病等癲癇疾病。臨床研究發現,基因突變是導致癲癇的重要原因,EOEE疾病發作時腦部異常放電現象與腦神經元的分化遷移、神經遞質的合成釋放過程、突觸的產生修剪以及細胞膜的結構等多種基因有關,因而檢測相關基因異常尤為重要[6,7]。康慶云[8]等在研究癲癇疾病患兒SCNIA基因突變時發現,鈉通道蛋白編碼基因(Sodium Voltage-Gated Channel Alpha Subunit 1,SCN1A )與Dravet綜合征發作有關,可通過細胞內外物質交換過程干擾基因編碼鈉通道α亞基,致使患兒全身癲癇發作,因而需對尚未明確基因缺陷的EOEE患兒進行SCNlA基因檢測。此外,細胞周期依賴激酶樣5(cyclin-dependent kinase-like 5,CDKL5)被認為與EOEE患兒早發性驚厥有關,主要表型為嬰兒痙攣癥、Hanefeld型不典型Rett綜合征和孤獨癥障礙,可通過影響遺傳基因表達致使患者發育阻滯和智力損傷[9]。
本實驗研究已在我院確診并接受治療的40例早發性癲癇腦病患兒,共發現32例(80.00%)SCNIA、CDKL5基因突變,其中男12例,女28例,疾病發作并接受診療時間分別為出生6d至18個月(10.11±4.29)個月。在32例基因突變患兒中,嬰兒痙攣癥患兒有23例,非特異性EOEE患兒10例,Dravet綜合征4例,大田原綜合征患兒3例,在發現的突變類型中,有17例(42.50%)錯義突變,3例(7.50%)插入突變,3例(7.50%)無義突變,3例(7.50%)剪切位點突變,6例(15.00%)微小缺失突變。經排查發現,所有患兒的父母均非近親婚配生育,且家族內未發現癲癇或神經疾病史。這說明,該32例患兒的SCNIA、CDKL5基因突變均為新生突變,且EOEE患兒癲癇發作類型多樣,發育落后,需及早完善基因篩查[10]。在王翠[11]等對Dravet綜合征患兒SCNIA基因突變特點的研究結論中,患兒大多出現無熱驚厥、肌陣攣和行為運動障礙,與梅道啟[12]等研究CDKL5基因突變出現的驚厥、目光對視無神、身體生長緩慢、手足動作刻板等臨床癥狀相似。在本實驗中,32例SCNIA、CDKL5基因突變患者存在不同程度的智力發育落后情況,尚無患兒具備有意義語言和獨立行走能力,21例(65.63%)患兒獨坐時間明顯低于同年齡正常兒童,28例(87.50%)患兒具有孤獨癥征兆,11例(34.38%)存在手足功能低刻板和身體發育緩慢兩組基因突變類型患兒均出現不同程度的驚厥、強直陣和全身痙攣,與上述研究結果一致。該實驗結果表明,患兒在發病階段可出現兩種以上癲癇發作類型,且可能由兩個或兩個以上基因突變造成。此外,本實驗32例SCNIA、CDKL5基因突變患兒中,SCNIA基因突變17例(42.50%),1CDKL5基因突變5例(37.50%),其中男11例(SCNIA基因突變5例,CDKL5基因突變6例),女21例(SCNIA突變12例,CDKL5突變9例)。從該結果中可發現,SCNIA、CDKL5基因突變中女性患兒數量要多于男性,且女性CDKL5基因突變在兩種基因突變中的比例要大于男性。初步分析,女性CDKL5突變率大于男性可能是受到染色體影響,與男性的一條X染色體相比,女性具有兩條。當其中一條X染色體基因突變時,另一X染色體基因起到起代償作用,使得女性僅發生錯義突變而不致病。但是,本實驗主要目的是探究EOEE患兒SCN1A、CDKL5基因突變的臨床特征,為獲取不同性別之間的SCN1A、CDKL5基因突變差異,還需擴大樣本做進一步分析。
綜上所述,SCN1A、CDKL5基因突變相關的EOEE疾病在患兒群體中發病形式多樣,嚴重影響患兒的生長發育過程,需在早期抗癲癇藥物和食療基礎上探索更加針對性的治療方法,以此對癥治療。此外,男女不同性別所導致的SCN1A、CDKL5基因突變致病機制有可能存在差異,為獲取更加具體、真實的研究數據還需大樣本做進一步分析。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國民間療法(2021年5期)2021-06-09 09:21:04
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
飲食科學(2017年5期)2017-05-20 17:11:53
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
安徽醫科大學學報(2015年9期)2015-12-16 11:09:44
西南軍醫(2015年4期)2015-01-23 01:19:30
中國中醫藥現代遠程教育(2014年20期)2014-03-01 04:31:21
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
