miR-216a、miR-301a調控BMPR2促進缺氧環境下肺動脈平滑肌細胞的增殖
2022-05-31 04:24:32林瀟鄭煒平李鴻茹陳愉生周夢
江蘇大學學報(醫學版) 2022年3期
林瀟,鄭煒平,李鴻茹,陳愉生,周夢
(福建省立醫院 1. 老年科; 2. 呼吸與危重癥醫學科; 3. 風濕免疫科,福建 福州 350001)
肺動脈高壓(pulmonary arterial hypertension, PAH)是由多因素引起的肺血管阻力增加,引發肺動脈壓力異常升高的臨床綜合征,其病理生理特點主要為肺動脈平滑肌細胞(pulmonary arterial smooth muscle cells, PASMC)過度增殖、肥大,后期逐漸演變為肺血管叢狀纖維化[1],右心室后負荷增加,最終導致右心功能衰竭[2]。目前PAH的治療主要包括一氧化氮、內皮素受體拮抗劑和前列環素類似物[3],但總體效果不佳,死亡率高,因此亟需尋找新的分子通路靶點用于藥物研發。當前微小RNA(miRNA)參與PAH發病機制的研究已成為學術界的熱點,多篇文獻已報道,miR-21、miR-124、miR-140和miR-663等多種miRNA參與調控PASMC的增殖、遷移[4-7],有研究發現miR-216a、miR-301a在PAH大鼠或人的PASMC中表達上調[8-9],然而缺氧環境下miR-216a、miR-301a調控PASMC增殖的相關機制研究較少。本研究通過體外細胞實驗探討miR-216a、miR-301a對缺氧條件下人PASMC(HPASMC)增殖的影響及相關分子機制,旨在為治療PAH開發新型靶向藥物提供理論基礎。
1 材料與方法
1.1 主要材料
HPASMC及iCell原代平滑肌細胞基礎培養基(上海賽百慷生物技術公司);超純RNA提取試劑盒、超純miRNA提取試劑盒及BCA蛋白定量試劑盒(北京康為世紀生物科技公司);mRNA逆轉錄試劑盒、miRNA逆轉錄試劑盒及miRNA熒光定量qPCR檢測試劑盒(南京諾唯贊生物科技公司);mRNA熒光定量qPCR檢測試劑盒(廈門生命互聯科技公司);CCK8細胞增殖檢測試劑盒(江蘇凱基生物技術公司);RIPA細胞裂解液(北京普利萊基因技術公司);ECL超敏發光液(美國賽默飛公司);骨形成蛋白2型受體(bone morphogenetic protein receptor type 2, BMPR2)、增殖細胞核抗原(proliferating cell nuclear antigen, PCNA)、β-肌動蛋白抗體及辣根過氧化物酶標記的二抗(英國Abcam公司);Lipofectamine 3000轉染試劑(美國Invitrogen公司);雙熒光素酶報告檢測試劑盒(上海碧云天生物技術公司);293T細胞(河北北納生物科技公司);miR-216a模擬物、miR-216a抑制物(anti-miR-216a)、miR-301a模擬物、miR-301a抑制物(anti-miR-301a)、模擬物對照(miR-NC)及抑制物對照(anti-miR-NC)、BMPR2過表達質粒(pcDNA-BMPR2)及空白載體(pcDNA)均購自江西中洪博元生物公司。
1.2 主要方法
1.2.1 細胞培養 將HPASMC加入iCell原代平滑肌細胞基礎培養基中,常氧組細胞于21% O2、5% CO2、74% N2的37 ℃恒溫培養箱中培養48 h,缺氧組細胞于3% O2、5% CO2、92% N2的37 ℃培養箱中培養,根據HPASMC在缺氧環境下培養時間的不同分為3組,分別為缺氧12 h、缺氧24 h和缺氧48 h組。
1.2.2細胞轉染 HPASMC傳代培養至細胞密度達70%時,更換為無血清的培養基,根據Lipofectamine 3000試劑盒說明書,將miR-216a和miR-301a模擬物、anti-miR-216a、anti-miR-301a、miR-NC、anti-miR-NC、pcDNA-BMPR2及空白載體轉染入HPASMC,轉染4 h后加入血清含量為20%的完全培養基,放回缺氧培養箱中繼續培養48 h后進行檢測。
1.2.3 qRT-PCR 提取常氧組和各缺氧組HPASMC中的BMPR2 mRNA和miR-216a、miR-301a,紫外分光光度計檢測RNA純度和濃度,分別通過mRNA和miRNA逆轉錄試劑盒合成cDNA,將cDNA分別采用mRNA和miRNA熒光定量qPCR檢測試劑盒進行擴增,反應步驟為預變性95 ℃,10 min;變性95 ℃,10 s;退火58 ℃,30 s;延伸72 ℃,30 s;40個循環。分別以β-肌動蛋白和U6作為mRNA和miRNA的內參,miR-216a、miR-301a、BMPR2 mRNA的相對表達量根據2-△△Ct法計算。miR-216a上游引物:5′-CGCGTCACAGTGGTCTCTGG-3′,下游引物:5′-AGTGCAGGGTCCGAGGTATT-3′;miR-301a上游引物:5′-CGCGCAGTGCAATAGTATTGT-3′,下游引物:5′-AGTGCAGGGTCCGAGGTATT-3′;BMPR2上游引物:5′-TTGCCGTCTTGCTCATTCT-3′,下游引物:5′-GTCTATTTCCAGTCAGCCTCAT-3′;β-肌動蛋白上游引物:5′-TGGCACCCAGCACAATGAA-3′,下游引物:5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′;U6上游引物:5′-CTCGCTTCGGCAGCACA-3′,下游引物:5′-AACGCTTCACGAATTTGCGT-3′。
1.2.4 CCK8檢測HPASMC增殖活力 將造模的HPASMC消化、重懸、計數、鋪板,細胞密度為5×103個/孔,將細胞培養48 h后,待測的96孔板細胞換成相同的培養基,每孔加入10 μL CCK8試劑,置于培養箱中孵育2 h,酶標儀在450 nm波長處檢測每孔的光密度值,并計算常氧和不同缺氧時間培養下HPASMC的存活率,以及缺氧環境下轉染miR-216a或miR-301a抑制物,同時轉染miR-216a或miR-301a聯合pcDNA-BMPR2后HPASMC的存活率。
1.2.5 蛋白免疫印跡檢測BMPR2、PCNA表達 使用RIPA細胞裂解液提取HPASMC中的蛋白,BCA試劑盒測定蛋白濃度。將蛋白質加熱變性后,進行十二烷基苯磺酸鈉凝膠電泳2 h,后用300 mA恒流轉至PVDF膜80 min。將膜浸入5%脫脂奶粉封閉液中孵育2 h,4 ℃下與BMPR2抗體、PCNA抗體及β-肌動蛋白抗體孵育過夜,二抗溶液中室溫孵育2 h。在PVDF膜上滴加ECL發光液,并在凝膠成像系統中曝光。采用Image-Pro軟件分析各抗體條帶灰度值。
1.2.6 雙熒光素酶報告檢測各轉染組螢火蟲熒光素酶和海腎熒光素酶活性 將含有miR-216a結合位點的BMPR2 3′-UTR的野生型(WT)和突變型(MUT)片段克隆到pmirGLO質粒上,分別構建BMPR2野生型熒光素酶載體(BMPR2 WT-1)和BMPR2突變型熒光素酶載體(BMPR2 MUT-1),用同樣的方法構建與miR-301a結合位點互補的BMPR2 WT-2載體和BMPR2 MUT-2載體。將293T細胞鋪板,待細胞完全貼壁后密度達70%時,將miR-216a、miR-301a、miR-NC和構建的BMPR2熒光素酶載體共轉染293T細胞。轉染48 h后裂解細胞,通過雙熒光素酶檢測法分別檢測各轉染組螢火蟲熒光素酶活性和海腎熒光素酶活性,驗證BMPR2與miR-216a、BMPR2與miR-301a之間的關系。
1.3 統計學分析
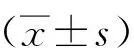
2 結果
2.1 缺氧對細胞增殖活力及miR-216a、miR-301a、BMPR2表達的影響
CCK8結果顯示,HPASMC的增殖活力隨缺氧時間延長逐漸增加,Spearman相關性分析提示細胞增殖活力與缺氧時間呈正相關(r=0.865,P<0.001)。因為在本研究中缺氧48 h組細胞增殖活力最強,故后續實驗選擇缺氧48 h進行。qRT-PCR結果顯示,miR-216a在缺氧24 h和48 h的表達水平均顯著高于常氧組及缺氧12 h組(P<0.05),miR-216a表達與缺氧時間也呈正相關(r=0.870,P<0.001)。miR-301a在缺氧12 h、24 h和48 h組的表達量均高于常氧組(P<0.05),并且在缺氧24 h組表達量最高。BMPR2 mRNA的表達水平隨著缺氧時間延長而逐漸降低,兩者呈負相關(r=-0.978,P<0.001)。見圖1。

①:常氧組,②:缺氧12 h組,③:缺氧24 h組,④:缺氧48 h組。a:P<0.05,與常氧組比較;b:P<0.05,與缺氧12 h組比較;c:P<0.05,與缺氧24 h組比較
2.2 缺氧環境下miR-216a、miR-301a對HPASMC增殖的影響
CCK8和蛋白免疫印跡結果顯示,與常氧組比較,缺氧組HPASMC的細胞增殖活力增強,PCNA表達增加(P均<0.05),轉染抑制物對照miR-NC幾乎不影響缺氧組HPASMC的增殖活力及PCNA表達。轉染miR-216a或miR-301a抑制物后,HPASMC增殖活力受到抑制,PCNA表達下降(P均<0.05),提示下調miR-216a或miR-301a抑制缺氧環境下HPASMC的增殖能力,間接表明缺氧條件下miR-216a、miR-301a能促進HPASMC的增殖(圖2)。

①:常氧組,②:缺氧組,③:缺氧+anti-miR-216a-NC組,④:缺氧+anti-miR-216a組,⑤:缺氧+anti-miR-301a-NC組,⑥:缺氧+anti-miR-301a組。a:P<0.05,與常氧組比較;b:P<0.05;與缺氧組比較;c:P<0.05,與缺氧+anti-miR-216a-NC組比較;d:P<0.05,與缺氧+anti-miR-301a-NC組比較
2.3 BMPR2是miR-216a、miR-301a的靶基因
借助Starbase數據庫(https://starbase.sysu.edu.cn)搜索發現miR-216a與BMPR2 mRNA的3′-UTR中的7個堿基互補,miR-301a也與BMPR2 mRNA的3′-UTR中的7個堿基互補,提示BMPR2可能是這兩個miRNA分子的靶點(圖3A)。雙熒光素酶報告結果顯示,miR-216a聯合BMPR2 WT-1轉染組的熒光素酶活性顯著低于miR-NC聯合BMPR2 WT-1轉染組和單純BMPR2 WT-1轉染組(P均<0.05),而miR-216a聯合BMPR2 MUT-1轉染組的熒光素酶活性與miR-NC聯合BMPR2 MUT-1轉染組和單純BMPR2 MUT-1轉染組比較無顯著差異(圖3B),提示BMPR2是miR-216a的靶基因。同樣,miR-301a聯合BMPR2 WT-2轉染組的熒光素酶活性顯著低于miR-NC聯合BMPR2 WT-2轉染組和單純BMPR2 WT-2轉染組(P均<0.05),miR-301a聯合BMPR2 MUT-2轉染組的熒光素酶活性與miR-NC聯合BMPR2 MUT-2轉染組和單純BMPR2 MUT-2轉染組比較無顯著差異(圖3C),提示BMPR2也是miR-301a的靶基因。

A:Starbase數據庫預測BMPR2分別與miR-216a、miR-301a的結合位點;B:雙熒光素酶報告測定BMPR2與miR-216a之間的關系;C:雙熒光素酶報告測定BMPR2與miR-301a之間的關系。①:BMPR2 WT-1組,②:BMPR2 WT-1+miR-NC組,③:BMPR2 WT-1+miR-216a組,④:BMPR2 MUT-1組,⑤:BMPR2 MUT-1+miR-NC組,⑥:BMPR2 MUT-1+miR-216a組,⑦:BMPR2 WT-2組,⑧:BMPR2 WT-2+miR-NC組,⑨:BMPR2 WT-2+miR-301a組,⑩:BMPR2 MUT-2組,:BMPR2 MUT-2+miR-NC組,:BMPR2 MUT2+miR-301a組。a:P<0.05,與BMPR2 WT-1組比較;b:P<0.05,與BMPR2 WT-1+miR-NC組比較;c:P<0.05,與BMPR2 WT-2組比較;d:P<0.05,與BMPR2 WT-2+miR-NC組比較
2.4 缺氧環境下miR-216a、miR-301a下調BMPR2基因促進HPASMC的增殖
蛋白免疫印跡結果顯示,與常氧組比較,缺氧組BMPR2表達降低(P<0.05),轉染了pcDNA-BMPR2后,HPASMC中的BMPR2表達升高(P<0.05)。與轉染miR-NC聯合pcDNA-BMPR2組相比,當轉染了miR-216a或miR-301a聯合pcDNA-BMPR2后,BMPR2明顯下降(P<0.05),提示miR-216a或miR-301a抑制BMPR2的表達。與缺氧環境下轉染空白載體比較,缺氧環境下轉染pcDNA-BMPR2,CCK8結果顯示細胞增殖活力降低(P<0.05),蛋白免疫印跡結果顯示HPASMC中的PCNA表達下降(P<0.05),提示BMPR2抑制HPASMC的增殖。與轉染miR-NC聯合pcDNA-BMPR2組比較,當轉染miR-216a或miR-301a聯合pcDNA-BMPR2后,細胞增殖活力明顯升高(P<0.05),PCNA表達也呈上升趨勢,但差異無統計學意義。綜上所述,缺氧環境下miR-216a或miR-301a通過抑制BMPR2,促進HPASMC的增殖。見圖4。
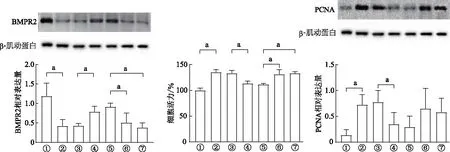
①:常氧組,②:缺氧組,③:缺氧+pcDNA組,④:缺氧+pcDNA-BMPR2組,⑤:缺氧+miR-NC+pcDNA-BMPR2組,⑥:缺氧+miR-216a+pcDNA-BMPR2組,⑦:缺氧+miR-301a+pcDNA-BMPR2組。a:P<0.05
3 討論
缺氧是引發PAH的重要因素之一。目前有關慢性缺氧引發PAH的機制尚未完全闡明,有研究表明缺氧可能通過促進大鼠PASMC中的煙酰胺腺嘌呤二核苷酸磷酸氧化酶4表達增加,誘導活性氧的產生,促進PASMC增殖[10]。新近研究發現缺氧還可誘導膠質瘤相關癌基因蛋白-1的異常表達,通過調控PASMC的增殖和焦亡過程,促進PAH的形成[11]。本研究結果顯示,HPASMC的增殖活力隨缺氧時間延長而逐漸增加,HPASMC在缺氧組中的PCNA表達水平高于常氧組,均提示缺氧環境能顯著誘發肺血管平滑肌細胞增殖,促進肺血管重塑。
已有研究發現miR-130/301家族在HPASMC中表達上調,并通過抑制PPARγ表達,調控下游STAT3/miR-204信號通路,促進HPASMC增殖[9]。另有研究表明miR-130/301家族受轉錄調節因子YAP/TAZ激活,促進肺血管細胞外基質的重塑,參與PAH的形成[12]。目前有關miR-216a促進PAH形成機制的文獻報道極少,Chinnappan等[8]通過對HIV病毒轉基因大鼠注射可卡因以構建PAH模型,發現miR-216a在這類大鼠模型的PASMC中呈現高表達,進一步研究發現miR-216a通過沉默下游BMPR2基因表達,促進PASMC的增殖和遷移。而關于miR-216a在缺氧誘導PAH模型中的發病機制研究尚未見報道。本研究發現,隨著缺氧時間延長,miR-216a表達水平增加;miR-301a在缺氧24 h的表達水平也顯著高于常氧組和缺氧12 h組。通過下調缺氧環境中HPASMC的miR-216a和miR-301a表達,發現細胞增殖活力和PCNA表達均受到抑制,表明miR-216a和miR-301a可能參與PAH的發病過程。
BMPR2是轉化生長因子(transforming growth factor, TGF)超家族中一類重要的細胞因子,已被發現與肺動脈高壓的發病機制密切相關。Roberts等[13]報道BMPR2基因突變失活是特發性PAH的主要發病原因之一。Jiang等[14]發現在博萊霉素誘導PAH大鼠模型中,BMP9/BMPR2/SMAD信號傳導通路受到抑制,繼而導致肺微血管內皮細胞凋亡增加及肺血管平滑肌增厚。另有報道發現恢復BMPR2信號傳導能夠緩解甚至逆轉肺血管重塑[15]。本研究發現BMPR2 mRNA表達隨著缺氧時間延長而逐漸下降,對缺氧組HPASMC轉染pcDNA-BMPR2,細胞增殖活力明顯降低,PCNA表達下降,上述結果提示BMPR2具有抑制HPASMC增殖的作用。
目前,miRNA參與調節BMPR2基因或BMP/TGF-β信號通路誘導PAH形成機制的研究已成為學者廣泛關注的熱點。例如miR-17-92系列通過IL-6/STAT3信號通路下調BMPR2表達,對PASMC和肺血管內皮細胞發揮促增殖和抗凋亡作用[16]。miR-424/503通過上調Apelin基因,恢復BMPR2信號傳導,抑制PASMC和肺血管內皮細胞增殖[17]。新近研究發現miR-182通過沉默髓系相關分化標志基因(myeloid-associated differentiation marker, MYADM),促進BMP/TGF-β信號通路傳導,延緩缺氧誘導PAH的發病進程[18]。本實驗通過雙熒光素酶報告測定證實BMPR2是miR-216a、miR-301a的下游靶點。進一步研究表明,同時轉染miR-216a或miR-301a聯合pcDNA-BMPR2可顯著抑制BMPR2表達水平,細胞增殖活力增強,PCNA表達上調,提示miR-216a、miR-301a通過抑制BMPR2參與缺氧誘導PAH的形成。
綜上所述,缺氧條件下miR-216a和miR-301a均能促進HPASMC增殖,其作用機制與抑制下游BMPR2表達有關。本研究在設計上還存在以下不足:首先,本研究僅通過體外細胞實驗來探討miRNA參與PAH發病的相關機制,尚缺少動物實驗及臨床試驗的驗證;其次,PAH的發病過程不僅與PASMC的增殖有關,也和細胞的遷移、凋亡等多種生物學行為有關聯。故今后有必要進一步探討上述兩個miRNA對缺氧環境下PASMC遷移及凋亡的影響,并研究潛在的信號傳導通路,同時增加動物體內實驗及臨床試驗來確認miR-216a和miR-301a在PAH發病中的作用。另外,近年來環狀RNA(circRNA)與PAH關系的研究已成為學者關注的前沿熱點,已發現circRNA可通過吸附miRNA(miRNA海綿作用),削弱miRNA對下游靶基因的抑制,上調靶基因的表達,參與疾病的發病過程[19]。今后可在本研究基礎上繼續探尋miR-216a、miR-301a上游可能參與PAH發病的circRNA,探索相關分子通路,為未來精準化治療提供新的靶點。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
