不銹鋼酸洗廢混酸流化床焙燒再生特性的實驗研究
2022-05-26 03:00:30徐勁松林敏陳曉平馬吉亮耿鵬飛鮑學兵劉道銀梁財
化工學報 2022年5期
關鍵詞:煙氣
徐勁松,林敏,陳曉平,馬吉亮,耿鵬飛,鮑學兵,劉道銀,梁財
(1 能源熱轉化及其過程測控教育部重點實驗室,東南大學能源與環境學院,江蘇南京 210096;2 廣州市中綠環保有限公司,廣東廣州 510670)
引 言
不銹鋼在冷軋和退火過程中,表面由于氧化作用會形成一層金屬氧化物層(氧化鐵、氧化鉻和其他氧化物)[1],為保證不銹鋼的表面質量、使用壽命和加工精度[2],必須采用酸洗工藝將不銹鋼表面的氧化物層去除。根據不同的不銹鋼品種,采用不同的酸洗介質,如硫酸、鹽酸、混酸(硝酸、氫氟酸)。目前,不銹鋼酸洗普遍采用混酸(HNO3+HF)作為酸洗介質[3]。
混酸酸洗工藝的主要化學反應如下:
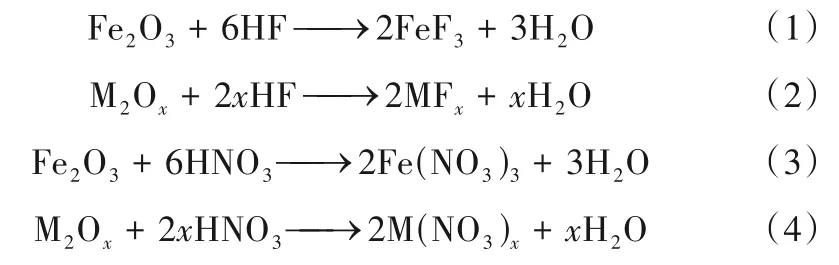
M 為不銹鋼表面氧化物層中的其他金屬元素,包括Cr、Mg、Ni、Mn、Mo、Ca等。
為保證不銹鋼酸洗效果,需不斷補充新酸,同時排出部分廢酸,因此酸洗工藝會產生大量的廢酸。由于廢混酸的pH 較低,且含有大量酸根離子和重金屬離子,直接排放既會對水質和土壤環境造成嚴重污染,也是一種資源浪費[4-8]。為防止廢混酸的污染并實現酸和金屬元素的有效回收,研究機構和工業界開展了大量的研究和工程探索,并已開發出部分較為成熟的技術。
朱冰[4]采用黃鈉鐵礬法和萃取法對廢混酸進行協同處理,發現廢混酸中金屬元素的回收率較高(鐵、鉻和鎳的回收率分別達到92.82%、89.14%和92.71%),但無法同步回收HF 和HNO3。王超[9]采用Scanacon 酸回收法對廢混酸進行處理,發現該方法可同步回收游離酸,降低硝酸和氫氟酸的消耗,并減少酸污泥沉淀雜質的形成,使廢棄物排放量降低50%,但該方法不能實現金屬的同步回收。
可同時回收酸和金屬離子的方法包括離子交換樹脂法、雙極膜電滲析法和焙燒法。王貴喜等[10-11]采用離子交換樹脂法對廢混酸進行處理,發現HF 和HNO3的回收率分別為86%和90%。不同的離子交換樹脂會產生不同的效果,李菲等[12-13]利用強酸性陽離子交換樹脂吸附廢混酸中的鉻和鐵,發現樹脂對Cr3+和Fe3+的飽和吸附回收量較高(60.34 mg/g 和65.3 mg/g)。李小明等[14-17]對雙極膜電滲析法、焙燒法進行研究,發現雙極膜電滲析法對硝酸、氫氟酸和金屬鹽的回收率分別為90%、65%和50%,但是膜易被K2SiF6污染,成本高;焙燒法對硝酸、氫氟酸的回收率分別為75%和98%,對金屬鹽的回收率為82%~90%,但該方法運行費用較高,裝置復雜。
焙燒法的綜合回收率最高,也最環保。焙燒法分為噴霧焙燒法和流化床焙燒法,噴霧焙燒法的產物是氧化鐵粉末,流化床焙燒法的產物是氧化鐵顆粒[18]。Nandy 等[19]采用流化床焙燒法對廢鹽酸進行處理,研究發現HCl 和Fe2O3的回收率分別達到99%和98%。廢鹽酸流化床焙燒法處理技術的高回收率,為廢混酸的處理提供了新思路,但目前還未見采用流化床焙燒法處理廢混酸(HNO3+HF)的研究和工業應用的報道。
本文借鑒廢鹽酸流化床焙燒法,開展廢混酸(HNO3+HF)流化床焙燒技術研究,重點考察流化床密相區溫度和初始床料粒徑對廢混酸中酸的再生與金屬離子在床料表面附著生長的影響規律,并對操作參數進行優化,為廢混酸流化床焙燒技術的工程應用提供理論參考和基礎數據。
1 實驗材料和方法
1.1 實驗系統
自行構建的廢混酸流化床焙燒法再生實驗系統如圖1所示,主要包括流化床反應爐、尾氣處理系統、電加熱系統、噴酸系統、配氣系統和煙氣取樣及分析系統6個部分。
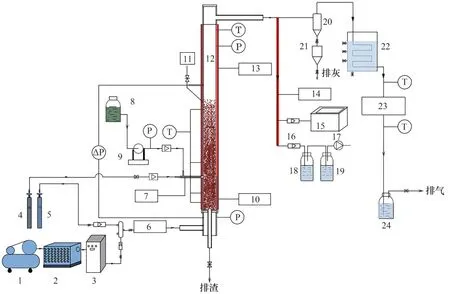
圖1 廢混酸流化床焙燒法再生實驗系統示意圖1—空氣壓縮機;2—冷干機;3—制氮機;4—氮氣鋼瓶;5—氧氣鋼瓶;6—預熱器;7—噴槍;8—廢混酸罐;9—電磁泵;10—密相區電加熱器;11—料罐;12—流化床反應爐;13—稀相區電加熱器;14—電加熱帶;15—煙氣分析儀;16—流量計;17—真空泵;18—去離子水;19—NaOH溶液;20—旋風分離器;21—灰斗;22—冷卻器;23—布袋除塵器;24—堿洗裝置Fig.1 Schematic diagram of the experimental system for regeneration of waste mixed acid by fluidized bed method1—air compressor;2—cold dryer;3—nitrogen generator;4—nitrogen cylinder;5—oxygen cylinder;6—preheater;7—spray gun;8—waste mixed acid tank;9—electromagnetic pump;10—electric heater in the dense phase zone;11—canister;12—fluidized bed reactor;13—electric heaters in the thin phase zone;14—electric heating belt;15—flue gas analyser;16—flow meter;17—vacuum pump;18—deionised water;19—NaOH solution;20—cyclone separator;21—grey bucket;22—cooler;23—bag filter;24—alkaline washing unit
(1)流化床反應爐:爐體由密相區和稀相區構成,密相區內徑為80 mm,高度為400 mm;稀相區內徑為100 mm,高度為1300 m,固定床高為40 cm。
(2)尾氣處理系統:包括旋風分離器、煙氣冷卻器、布袋除塵器和酸性氣體堿洗塔。
(3)電加熱系統:包括配氣預熱器、密相區加熱器、稀相區加熱器、流化風進氣管線加熱器和煙氣取樣管路加熱。
(4)噴酸系統:主要包括廢混酸罐、電磁泵、流量計和噴槍,廢酸噴槍采用雙流體霧化噴槍,且噴槍位于布風板上方20 cm 處,噴槍出口與爐壁內壁面齊平。
(5)配氣系統:主要包括空壓機、冷干機、制氮機、玻璃轉子流量計。
(6)煙氣取樣及分析系統:利用去離子水吸收煙氣中的HF,煙氣取樣流量為40 ml/min,取樣時間為10 min,采用離子色譜法檢測吸收液中的HF 濃度,并對時間進行積分得到HF 生成量。利用煙氣分析儀(MRU VARIO plus)在線檢測煙氣中的NOx濃度,并對時間進行積分得到NOx生成量。煙氣取樣點位于旋風分離器與煙氣冷卻器之間,取樣管路裝有加熱裝置和保溫層,管路溫度控制在230℃。
1.2 實驗材料
實驗用廢混酸由江蘇揚州某不銹鋼酸洗企業提供,廢混酸的化學組成通過離子色譜儀(Metrohm 930)分析得到,如表1所示。
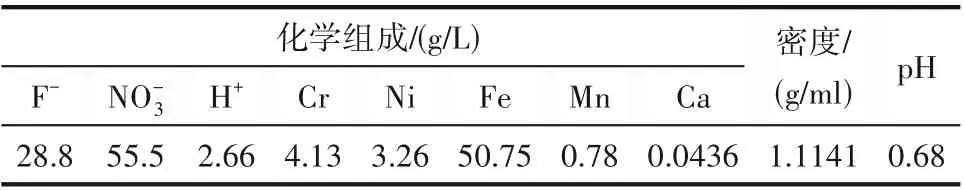
表1 廢混酸理化特性和成分分析Table 1 Physicochemical characteristics and composition analysis of waste mixed acid
實驗用氧化鐵顆粒由石家莊華邦礦產品有限公司制備,主要成分為Fe2O3(質量分數66.6%)和Fe3O4(質量分數33.4%),通過振動篩篩分得到兩種不同粒徑范圍的窄篩分顆粒,平均粒徑分別為0.3 mm 和0.4 mm。兩種顆粒的粒徑分布利用激光粒度儀(Zetasizer 2000)分析獲得,如圖2 所示。元素分析通過XPS(Thermo escalab 250Xi)分析得出,如表2所示。
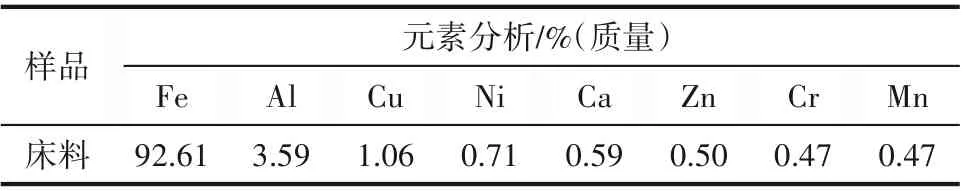
表2 氧化鐵床料元素分析Table 2 Elemental analysis of iron oxide bed material
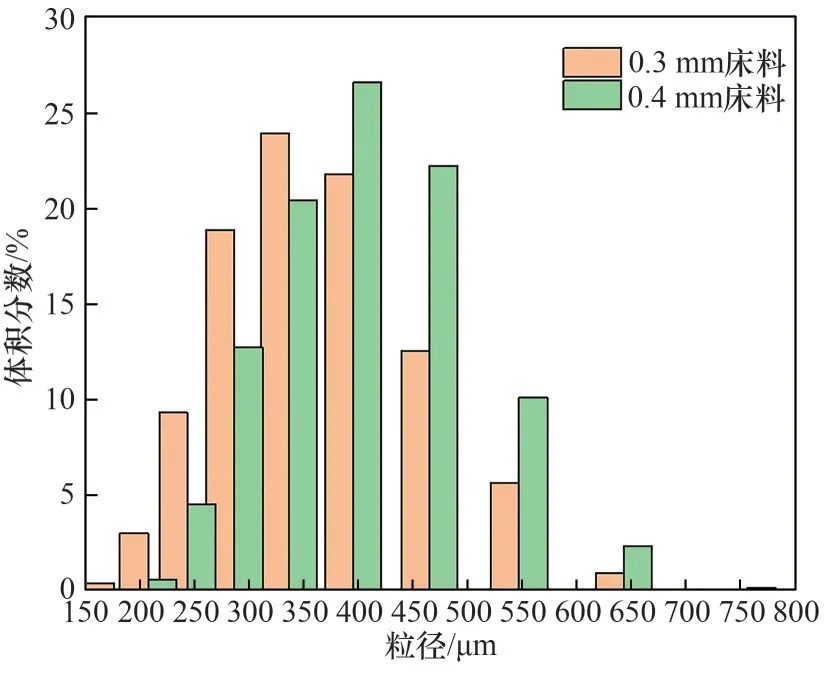
圖2 氧化鐵床料粒徑分布Fig.2 Iron oxide bed material particle size distribution
1.3 廢混酸流化床焙燒再生過程及實驗工況
1.3.1 廢混酸流化床焙燒再生過程 廢混酸焙燒前通常先進行加熱濃縮,去除其中的大部分游離酸和部分水分,然后噴入流化床反應爐。在反應爐內的高溫條件下,廢混酸中的硝酸鹽和氟酸鹽反應生成Fe2O3、M2Ox、HF、NO、NO2等產物,主要反應方程如下:
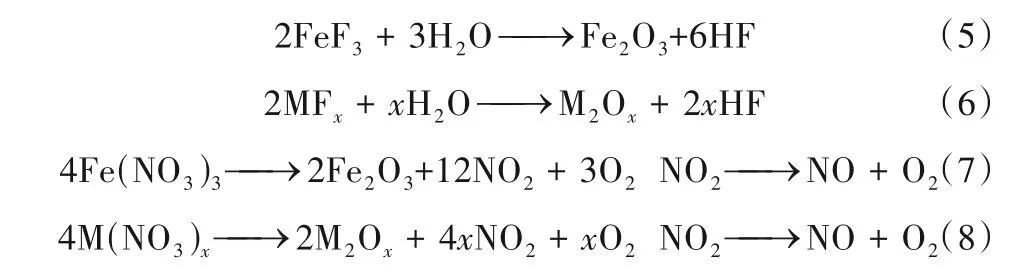
M 為廢酸中除鐵以外的其他金屬元素,包括Cr、Mg、Ni、Mn、Mo、Ca等。
上述反應中生成的細顆粒(主要為Fe2O3等金屬氧化物)附著在床料表面,隨床料從反應器底部排出并回收。煙氣從流化床反應爐的頂部排出,經旋風分離器后,煙氣中的金屬氧化物、未分解鹽和其他固體雜質顆粒被分離并重新送入流化床反應爐內。旋風分離器出口的高溫煙氣經文丘里冷卻器冷卻后再依次經過吸收塔和氧化塔(將NO 氧化為NO2),并采用工業新水對HF 和NO2進行回收再生,最后煙氣進入堿洗塔除酸后經煙囪排入大氣[20-21]。
1.3.2 實驗工況 本文主要研究流化床焙燒爐內的反應過程,重點考察流化床密相區溫度和初始床料粒徑對廢混酸中的酸與金屬離子再生的影響,因此未在尾部設置NO 氧化塔,酸回收效果基于尾部煙氣中NOx和HF的生成量進行考察。
研究表明,硝酸鐵在125℃時開始分解生成氧化鐵和NO2,NO2在150℃時開始分解為NO 和O2,硝酸鉻在447℃熱分解生成氧化鉻和NO2,NO2迅速分解[22],硝酸鎳和硝酸錳的分解溫度分別為573℃和473℃[23],FeF3的主要分解溫度高于600℃,因此實驗中密相區溫度均控制在650℃及以上。
實驗針對0.3 mm和0.4 mm初始平均粒徑床料,每種粒徑床料均進行密相區溫度設定值為650、700、750、800、850 和900℃熱態實驗。每個實驗工況下噴酸量均設為1.2 kg/h,噴酸持續時間為5 h,反應氣氛均為6%O2+94%N2(體積分數),初始床料添加量為6.5 kg。
2 實驗結果與討論
2.1 金屬氧化物在床料表面附著生長的變化規律
不同初始粒徑工況下床料的微觀形貌如圖3所示。密相區溫度為750℃時,未噴酸床料[圖3(a)、(c)]的表面平整,未出現細顆粒吸附。噴酸5 h 后,床料表面出現明顯的顆粒吸附[圖3(b)、(d)]。0.3 mm初始粒徑床料表面吸附的細顆粒保持類球狀形貌,尺寸均勻,表面平整,0.4 mm 初始粒徑床料表面吸附的顆粒大小不一,且相互粘連、表面粗糙。噴酸后顆粒表面微觀形貌的差異是由于流化風速的不同造成的。為保證兩種初始粒徑床料的流化狀態基本一致,0.3 mm 初始粒徑床料工況和0.4 mm 初始粒徑床料工況的流化速度分別為0.40 m/s 和0.52 m/s。流化速度的增加,導致流體對顆粒的曳力增大,使得黏結力較小的細顆粒不易在床料表面黏附;流化速度增加也使得細顆粒的揚析速率增加,細顆粒在床內的停留時間縮短,進一步降低了其在床料表面的吸附概率。同時,流化風速增加會加劇床料顆粒的碰撞磨損,因此吸附在床料表面但結合力較弱的細顆粒易脫離,只有黏結力較強的大顆粒仍然能吸附在床料表面,導致床料表面粗糙度增加。

圖3 床料表觀形貌變化(750℃下5 h后取樣分析)Fig.3 Changes in the apparent morphology of the bed material(analysis of samples taken after 5 h at 750℃)
為排除床料在流化狀態下發生磨損或者破碎產生細顆粒對實驗結果的影響,先進行了不噴酸時不同密相區溫度下流化5 h 的空白實驗,并利用激光粒度儀對空白實驗后的床料進行粒度分析。結果顯示,空白實驗中均未出現100 μm 以下的細顆粒,而在噴酸實驗時,床料中均有一定量的100 μm以下的細顆粒[圖4]。說明噴酸實驗條件下生成的細顆粒均為廢混酸在爐內反應的產物。
采用激光粒度分析儀測得不同密相區溫度和初始床料粒徑下,噴酸5 h 后床料的粒徑分布及累積分布,如圖4 所示。實驗溫度區間內(650~900℃),床料顆粒體積平均粒徑相對于初始床料粒徑均增大,密相區溫度從650℃升高到850℃,0.3 mm和0.4 mm 初始粒徑床料平均粒徑分別增加37.7 μm和39.4 μm,并達到峰值。同時床料中含有一定量100 μm 以下的細顆粒,當溫度分別超過850℃(0.3 mm 初始粒徑床料工況)和750℃(0.4 mm 初始粒徑床料工況)時,床料中細顆粒完全消失。霧化后的廢混酸液滴在高溫床料中經歷兩種過程:少量廢混酸液滴在床料表面附著和鋪展、分解生成的金屬氧化物晶體直接沉積在床料表面;大部分廢混酸液滴在床料間隙中快速蒸發,其中的硝酸鹽和氟化鹽分解生成金屬氧化物晶體顆粒和氣體產物,其中部分細顆粒會吸附在床料表面,剩余細顆粒則處于游離狀態[24-25],未附著在初始床料表面,在反應器內處于流化狀態。溫度較高、流化速度較大時,細顆粒不易在反應爐內停留,被氣體攜帶出爐內。
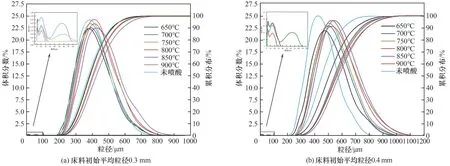
圖4 噴酸5 h床料粒徑分布及累積分布Fig.4 Particle size distribution of bed material for 5 h of acid spraying
不同初始粒徑和密相區溫度下,噴酸5 h,床料顆粒平均粒徑增加量和平均體積增加量分別如圖5和圖6所示。床料顆粒平均粒徑增加量和平均體積增加量為噴酸前后床料顆粒平均粒徑和平均體積之差。結合圖4,隨著密相區溫度的提高,床料的平均粒徑增加量有所增加,并在850℃時達到峰值,廢混酸的蒸發速率和金屬鹽的分解速率隨著密相區溫度的升高而加快,細顆粒生成速率增加,導致細顆粒在床料表面的吸附量增大,床料的平均體積增加量增加,850℃達到峰值。床料顆粒平均粒徑增加量和平均體積增加量達到峰值后繼續升高密相區溫度,由于流化風速的提高,使得細顆粒的運動速度加快,在床料表面更易反彈[26],導致細顆粒在床料表面的吸附程度有所下降。并且床料平均體積增加量隨床料初始粒徑的增大而增加。
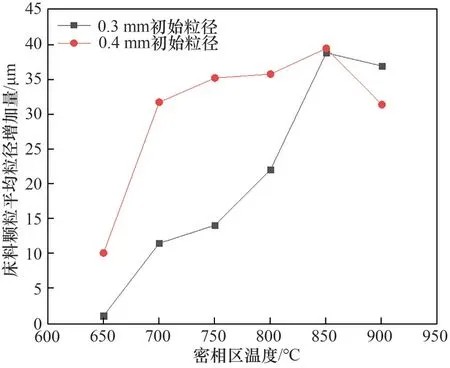
圖5 噴酸5 h床料顆粒粒徑增加量變化Fig.5 Change in particle size increase of bed material for 5 h of acid spraying
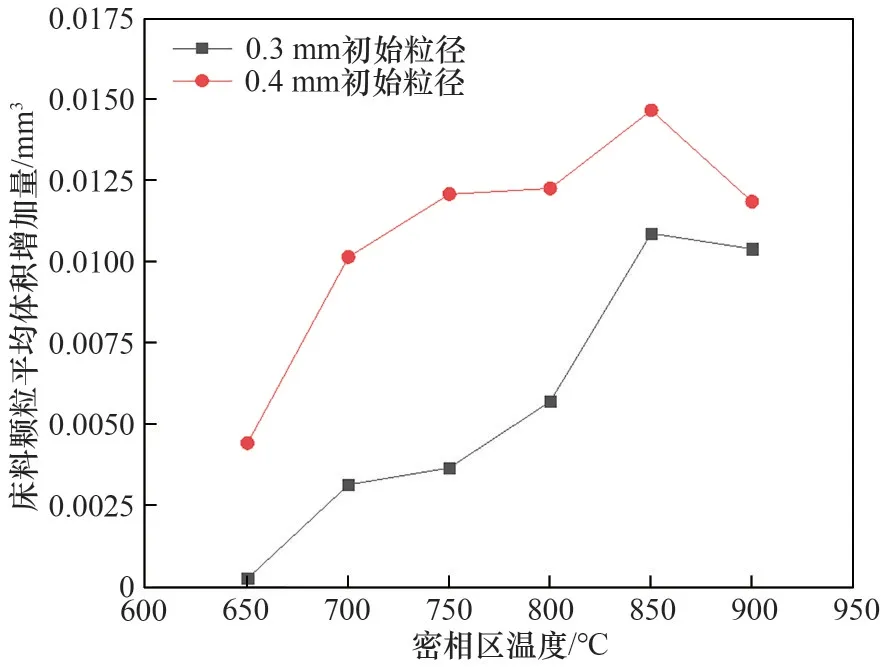
圖6 噴酸5 h床料顆粒平均體積增加量變化Fig.6 Change in the average volume increase of bed material particles for 5 h of acid spraying
利用X 射線光電子能譜儀(Thermo escalab 250Xi)分析得到不同初始床料粒徑和密相區溫度下床料顆粒表面主要金屬元素(Fe、Cr、Ni)含量變化,如圖7 所示。床料顆粒表面金屬元素檢測區域為400 μm×400 μm×10 nm。噴酸5 h后,隨著密相區溫度的升高,不同初始粒徑床料表面Fe元素占比均減小,Ni、Cr 元素占比逐步增大。因為廢混酸中相關金屬鹽的分解速率隨密相區溫度的升高而加快,使得細顆粒在床料表面吸附量增加。密相區溫度一定時,0.3 mm 床料表面Fe 元素占比均低于0.4 mm床料,這是由于生成物中Fe2O3、Cr2O3和NiO 的黏結力不同造成的。因為Cr2O3、NiO 和Fe2O3的熔點分別為2435、1984 和1565℃,所以在實驗溫度下Fe2O3的黏結力最強,最易吸附在床料表面。隨著床料初始平均粒徑增加,流化風速增加,使得黏結力較弱的Cr2O3和NiO 不易吸附在床料表面,造成床料表面Fe元素占比提高。

圖7 床料表面主要金屬元素含量占比變化Fig.7 Changes in the percentage of major metal elements on the surface of the bed material
2.2 NOx和HF生成量的變化規律
不同密相區溫度和初始床料粒徑下煙氣中NOx生成量的變化規律如圖8所示。隨著密相區溫度的升高,NOx的生成量顯著提高,在750℃達到峰值;溫度繼續升高,NOx生成量小幅下降。因為隨著密相區溫度的升高,廢混酸蒸發和硝酸鹽分解所需時間縮短[27],NOx生成量增加;當溫度到達750℃之后,硝酸鹽在爐內停留時間的影響將大于溫度對硝酸鹽分解速率的影響,NOx生成量有所下降。初始床料平均粒徑從0.3 mm 增大到0.4 mm 時,NOx生成量有所下降。0.4 mm初始粒徑床料工況中NO2生成量在750℃后高于0.3 mm 初始粒徑床料工況。因為初始床料平均粒徑增大,流化速度增加,硝酸鹽在反應器內停留時間縮短,硝酸鹽分解量以及NO2分解量小幅下降。
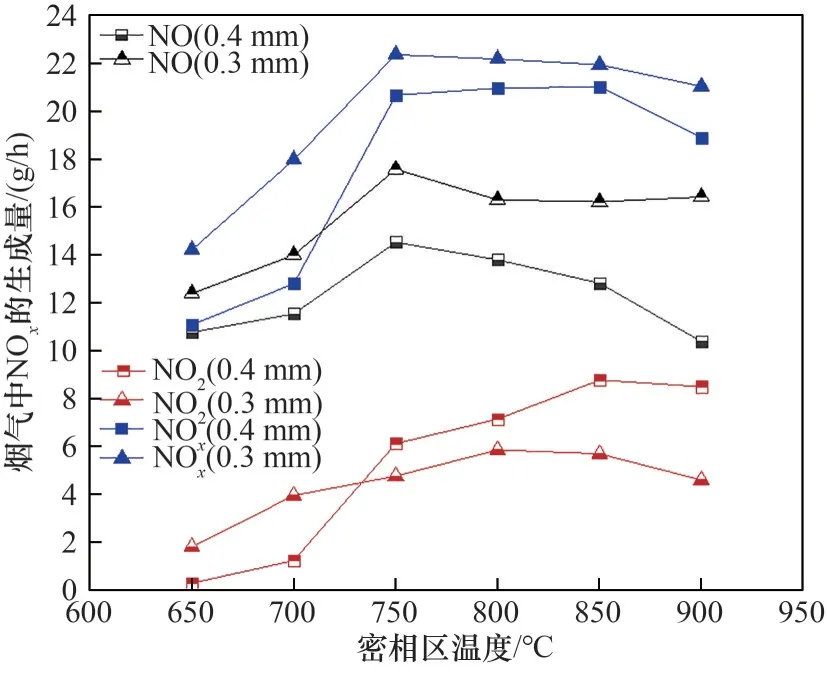
圖8 煙氣中NOx生成量與密相區溫度的關系Fig.8 Relationship between NOx generation in flue gas and temperature in dense phase zone
不同密相區溫度和初始床料粒徑下煙氣中HF生成量的變化規律如圖9 所示。煙氣中HF 生成量隨著密相區溫度的升高先增加后下降,不同初始床料粒徑對應的峰值溫度不同。氟化鹽的分解速率隨著密相區溫度的升高而加快,HF 的生成量增加,達到峰值以后,床料中的CaO 成分對HF 的化學固定效果提高,生成CaF2固體產物,如圖10 所示(以0.4 mm 初始床料粒徑850℃焙燒噴酸工況為例),CaF2分解溫度在1100℃以上。同時CaO固氟效果的最佳溫度隨操作參數的變化而變化[28-29],同時生成物中Fe、Mn 以及Al 的氧化物對HF 具有吸附或者共沉淀作用[30-34]。初始床料粒徑從0.3 mm 增大到0.4 mm 時,爐內流化速度增加,氟化鹽在反應爐內停留時間縮短,氟化鹽的分解量有所下降,HF 的生成量峰值溫度由750℃變化至800℃,并且反應生成的HF停留時間縮短,固氟量小幅下降[29,35]。
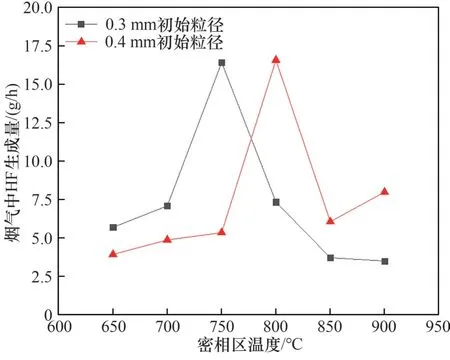
圖9 煙氣中HF生成量與密相區溫度的關系Fig.9 Relationship between HF generation in flue gas and temperature in dense phase zone
3 結 論
本文搭建了廢混酸流化床焙燒法再生實驗裝置并開展廢混酸(HNO3+HF)流化床焙燒實驗研究,分析了密相區溫度、初始床料粒徑對廢混酸中酸與金屬離子回收特性的影響規律,主要結論如下。
(1)增加密相區溫度能有效提高金屬氧化物在床料表面附著量,在850℃時附著量達到峰值,繼續升溫附著量小幅回落;煙氣中HF和NOx生成量也隨溫度升高而增加,并在750℃時達到峰值,繼續增加溫度,NOx生成量緩慢降低,而HF生成量大幅回落。
(2)受床料顆粒表面積的影響,隨著初始床料粒徑的增大,金屬氧化物在床料表面附著量提高。隨著流化風速的提高,HF 和NOx生成量有所降低,而NO2生成量在750℃后有所提高。
猜你喜歡
化工管理(2022年13期)2022-12-02 09:21:52
建材發展導向(2021年12期)2021-07-22 08:06:28
應用能源技術(2020年11期)2021-01-26 00:16:38
山東冶金(2019年2期)2019-05-11 09:12:16
測控技術(2018年2期)2018-12-09 09:00:52
電子測試(2018年1期)2018-04-18 11:52:15
當代化工研究(2016年9期)2016-03-20 16:22:15
中國資源綜合利用(2016年3期)2016-01-22 07:28:16
中國資源綜合利用(2016年2期)2016-01-22 07:27:41
有色金屬設計(2014年4期)2014-03-11 19:43:12
