纖毛通路基因罕見危害性基因變異對人腰骶部神經管畸形的致病性
2022-05-12 08:58:44蔡春泉陳曉麗
安徽醫科大學學報 2022年4期
汪 靜,劉 芳,謝 華,蔡春泉,姜 宏,陳曉麗
神經管畸形(neural tube defect,NTDs)是一類由于胚胎發育早期神經管閉合失敗或閉合不完全所致的嚴重出生缺陷。據統計,NTDs的全球發病率為0.5‰~2%,中國的發病率接近2.74‰[1]。NTDs是一類由環境和遺傳因素共同作用導致的復雜性疾病。目前通過人群及小鼠敲除模型研究已發現400多種可能導致神經管畸形的基因[2],其中熱點基因包括CELSR1、FZD6、PRICKLE1、DVL2、VANGL1、VANGL2等極性基因[3]。近年研究[4]認為新生突變是導致出生缺陷及神經發育疾病的常見原因。2015年,Lemay et al[5]對43個散發NTDs家系研究也證實新生突變是導致人腰骶部NTDs發生的原因之一。
纖毛是突出于細胞表面的微小細胞器并廣泛存在,與人類發育健康和疾病如麥克爾綜合征(Meckel syndrome,MKS)、Joubert綜合征(Joubert syndrome,JS)等密切相關。上述綜合征包括胎兒脊柱裂、腦脊膜膨出、腦組織缺如等與NTDs表型類似的臨床表現。但關于纖毛基因變異與人NTDs的相關性研究較少,本課題組前期研究[6]證實:同時參與纖毛發育的極性基因PARD3突變與人NTDs相關,且均為顱部NTDs。鮮有關于纖毛基因與人類腰骶部NTDs的相關性研究。該研究利用AmpliSeq技術設計了包括極性基因(如PARD3)及明確纖毛病候選基因在內的49個纖毛相關基因的編碼區靶向捕獲Panel,對56個散發腰骶部NTDs家系進行測序,以期明確纖毛罕見危害性變異對人腰骶部NTDs發生的致病性。
1 材料與方法
1.1 研究對象選擇在首都兒科研究所和天津市兒童醫院明確診斷的腰骶部神經管畸形患者56例,均無家族史,其中男性36例,女性20例。所有腰骶部神經管畸形患者由臨床醫師評估并影像學輔助診斷,最終手術明確診斷。全部患者父母亦由臨床醫師評估結合影像學輔助診斷無神經管畸形。本研究通過首都兒科研究所倫理委員會審查,所有患兒均由其父母或監護人簽署知情同意書。
1.2 主要儀器及試劑Qubit 2.0 熒光光度計、Ion OneTouch 油包水PCR儀、Ion OneTouch ES 儀、Ion Torrent PGM測序儀(美國Life Technologies 公司)。全血DNA提取試劑盒(美國Qiagen公司);Agencourt AMPure XP磁珠化試劑(美國Beckman Coulter公司);Qubit dsDNA HS Assay 試劑盒、Ion PGMTM200 Sequencing 測序試劑盒、Ion 316TM芯片、 Ion Xpress Barcode Adapters1-16試劑盒、Ion AmpliSeq Library 試劑盒(美國Life Technologies 公司)。
1.3 實驗方法
1.3.1DNA制備 采集56例神經管畸形患兒及其表型正常父母外周血提取基因組DNA,采用DNeasy Blood & Tissue kit試劑盒(美國Qiagen公司)提取DNA。取2 μl已提取的基因組DNA,應用紫外分光光度儀測量DNA濃度,A260/A280=1.8~2.0,A260/A230>2.0,計算基因組DNA含量。
1.3.2纖毛基因Panel的靶向深度測序 利用既往NTD小鼠模型中發現的纖毛基因[2]、OMIM系統內明確纖毛發育相關遺傳病(https://www.omim.org/)及課題組最近發表的文章[6]構建纖毛候選基因列表。采用Ion AmpliSeq Library在線設計系統形成纖毛基因的AmpliSeq靶向捕獲和多重PCR擴增試劑盒,之后和Ion Xpress Barcode Adapters試劑進行文庫構建,制備的文庫片段大小為200~300 bp;接著用Ion OneTouchTM200 Templete 試劑盒在Ion OneTouch上對文庫進行乳化,在Ion OneTouch ES上對磁珠進行富集;最后采用Ion PGM 200 試劑盒和Ion 316TM芯片在Ion Torrent PGM平臺進行測序。
1.3.3數據分析 實驗數據自動傳到Ion Torrent服務器,點擊已完成的測序,根據要求完成Coverage Analysis和Variant Caller分析,并對測序數據的數據量、平均測序深度、產生的BAM、BAI、VCF.GZ、XLS文件進一步分析。將VCF.GZ文件導入wANNOVAR(http://wannovar.usc.edu/ )軟件,分析所獲各變異位點在人群的分布頻率、氨基酸改變、變異分類(Indel、SNV),優先選擇Loss of Function變異,根據錯義變異的生物信息學軟件(Mutation Taster,PROVEAN,Polyphen-2,REVEL)預測結果確定其危害性。將BAM、BAI文件導入軟件IGV,明確各位點測序數據是否為真性突變,排除測序得到的假陽性數據。
2 結果
2.1 測序數據基本情況本研究收集的家系均為1個神經管畸形患兒配伍表型正常的父母,因此首選篩查雙位點罕見基因變異、新生基因變異。測序數據與公共數據庫[Genome1000、dbSNP、NHLBI、Exome Sequencing Project (ESP)、ExAC Browser]比對,結合神經管畸形的人群患病率,鎖定頻率小于1%的外顯子區非同義單核苷酸突變。對測序數據進行質量控制,測序覆蓋率97%~98%,平均測序深度400X~600X。
2.2 NTDs患者存在CRB2復雜雜合變異測序結果顯示,1例患兒存在CRB2基因兩個突變位點的復雜雜合變異(c.G1392C, p.R464S; c.T3448C, p.C1150R),且均為危害性突變。其中c.G1392C, p.R464S突變位點遺傳自患兒母親,公共數據庫中未見報道,另一個突變位點c.T3448C, p.C1150R遺傳自患兒父親,公共數據庫中為罕見變異(MAF<1%),見圖1、表1,兩突變位點在物種間高度保守,見圖2。

圖1 CRB2復雜雜合突變位點信息A:CRB2基因結構,紅色標注分別為患者攜帶CRB2;b:患者c.1392G>C變異來源于母親; c:患者c.3448T>C變異來源于父親;S、M、F分別代表患者、母親、父親

表1 突變位點基本信息
2.3 NTDs患者存在GLI3新生突變測序結果顯示,1例患兒存在GLI3基因的一個新生突變(c.C580T, p.H194Y),該突變經軟件預測為危害性突變,且公共數據庫均未見報道,見表1、圖3,該突變位點在物種間高度保守,見圖2。鑒于該家系發現的GLI3基因變異為新生錯義突變,筆者首先通過其他位點的連鎖分析證實了患者與父母的親子關系,用IGV查看家系測序數據,發現GLI3基因片段測序覆蓋率及測序深度較低,故采用Primer 3.0針對基因設計引物,對所有家系進行Sanger測序驗證,證實了GLI3新生突變位點的真實性,并且在其他55個家系均未見該基因的罕見變異,見圖4。其余54個神經管畸形家系尚未發現雙位點罕見基因變異及新生基因變異。

圖2 突變位點在8個物種間保守性分析
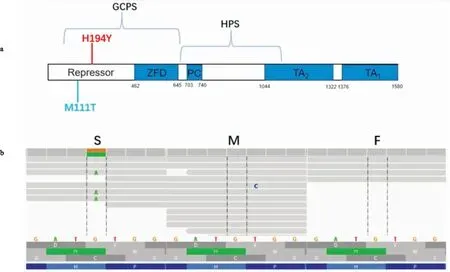
圖3 GLI3新生突變位點信息A:GLI3編碼蛋白結構,紅色標注為本文患者攜帶GLI3新生突變,藍色標注為文獻[7]中食管閉鎖伴有半椎體患者攜帶GLI3突變;b:panel測序結果顯示新生突變;S、M、F分別代表患者、母親、父親GLI3是GLI家族鋅指結構3。GLI家族鋅指蛋白是經典hedgehog(HH)信號通路的轉錄效應因子,參與胚胎形態發生的各個階段,并且是組織正常發育的必須調節因子。GLI3基因突變與格雷格頭、多指(趾)、并指(趾)綜合征(Greig cephalon polysyndactyly syndrome,GCPS)等相關。GLI3突變小鼠因大腦、脊柱、四肢、肺等多器官缺陷無法存活。2014年Yang et al[7]報道1例食管閉鎖伴半椎體患者攜帶一個GLI3的新生突變(M111T),進一步行蛋白功能實驗發現:該突變可導致內源性SKIL表達下降,并且GLI3與SKI家族蛋白-SKIL間的相互關系減弱,提示該突變可通過影響GLI3與SKIL的相互作用導致患者出現食管閉鎖及半椎體表型。2019年,Renard et al[14]首次發現一個GLI3基因SNP(c.4609C>T,rs35364414)在人類腰骶部NTDs中富集。研究顯示,人類腰骶部NTDs患者中存在GLI3的新生突變(H194Y),該突變同處于GLI3基因的Repressor域,該區域主要參與同SKI家族蛋白的相互作用,且GLI3基因M111T突變患者表型中存在半椎體,因此考慮GLI3基因的新生突變(H194Y)可能為該家系中患者的發病原因。后續仍需對H194Y突變進行功能試驗進一步驗證。

圖4 Q100家系成員GLI3基因變異Sanger測序驗證結果S:患者;M:母親;F:父親
3 討論
NTDs作為多基因疾病,存在高度遺傳異質性。絕大多數NTDs病因不明。纖毛在生物的胚胎發生和維持器官完整性中發揮重要作用。纖毛的功能異常會造成一系列表型復雜且嚴重的人類疾病稱為“纖毛病”。NTDs是一系列纖毛病的重要表型之一,如MKS患兒有腦膨出、多囊腎及并趾(指)等表型[8]。同時NTD小鼠也存在纖毛異常表型[3]。有研究[9]報道纖毛相關基因DNAAF1基因變異與人類NTDs發生相關。本課題組前期研究[10]顯示,纖毛基因的拷貝數變異與人類NTDs相關。
本研究利用AmpliSeq技術設計了包括纖毛極性基因(如PARD3)及明確纖毛病候選基因在內的49個相關基因的編碼區靶向捕獲Panel,對56個散發腰骶部NTDs家系進行測序,發現兩個可能與神經管畸形表型發生相關的基因:CRB2及GLI3。
CRB2與CRB1及CRB3為同源基因,均為Crumbs蛋白家族成員,在視網膜、大腦和腎臟中均有表達。Crumbs蛋白復合物(CRB-PALS1-PATJ或CRB-PALS1-MUPP1)與PAR蛋白復合物(PAR3-PAR6-aPKC)對發育的神經上皮細胞建立細胞極性有重要作用[11]。Boroviak et al[12]對CRB2基因的功能研究表明CRB2對維持其他極性細胞的穩定性也是必要的,無CRB2基因表達的胚胎干細胞在神經細胞開始分化時無法存活。人類CRB2基因突變可導致局灶性腎小球硬化(FGSG-9,MIM:616220)及一些纖毛相關疾病。本研究首次在NTDs家系發現CRB2基因突變,雖然其對神經管畸形的致病性尚不明確,但課題組前期通過病例對照研究[6]發現與CRB2基因密切作用的復合物——PARD3基因突變是人類NTDs的危險因素;通過突變子的體外細胞功能實驗和基因敲除的雞胚形態學實驗證實,PARD3基因aPKC結構域突變后可導致神經上皮細胞極性消失,該基因敲除后神經管腔化異常。小鼠模型研究[13]發現,CRB2基因敲除后,PALS1和PARD3基因表達明顯下調,而PALS1基因敲除后MDCK細胞的管腔化異常。本研究中1例單純腰骶部神經管畸形家系患者攜帶CRB2的復雜雜合變異,軟件預測均為危害性變異,考慮可能為該家系中患者的發病原因。尚需通過更多的病例-對照研究進一步證實CRB2基因罕見變異和人NTDs發生的關系。
