NMDA受體及其拮抗劑的研究進展
2022-05-11 06:54:50葉玉瑩羅揚文于沛
藥學研究 2022年4期
葉玉瑩,羅揚文,于沛
(暨南大學藥學院新藥研究所,廣東 廣州 510632)
作為神經遞質中的一種,谷氨酸受體在神經信號傳遞中扮演著重要的角色,其表達的數量、分布和種類等均影響著正常的神經生理功能,逆轉這些受體的功能變化對于治療或預防神經性疾病有重要意義。興奮性的突觸傳導主要通過激活兩類谷氨酸受體而實現,即離子型的谷氨酸受體(ionotropic glutamate receptors)和代謝型的谷氨酸受體(metabotropic glutamate receptors),根據對激動劑的親和力不同,分為3個亞型N-甲基-D-天冬氨酸(NMDA)受體、α-氨基-3-羥基-5-甲基-4-異亞唑丙酸(AMPA)受體和Kainate受體。本文重點關注NMDA受體研究進展,隨著研究的深入,以及研究儀器、方法和其他相關領域的發展,NMDA受體的結構逐漸明確,NMDA受體有多種亞型,由不同亞單位組成的受體亞型,具有不同的生物物理和生物化學特性。本文將從結構分布和生理活性這兩方面詳細總結NMDA受體各亞型的特點,介紹并匯總了目前研究較多的NMDA受體拮抗劑,綜合文獻的研究,提供該受體在神經性疾病中的重要作用,為相關研究提供信息依據。
1 NMDA受體的結構和分布
NMDA受體分布在全腦中,以海馬、大腦皮質、紋狀體、杏仁體為主。目前已鑒定出了多種NMDA受體亞基,包括廣泛表達的NR1,4個不同的NR2亞基(A、B、C、D),兩個NR3子單元(A和B)。
1.1 NR1 NR1是NMDA受體的基本亞基,是NMDA受體復合物的功能性亞單位,是實現該受體離子通道的功能所必需的,且NR1形成離子通道,是調節能力最強的神經遞質受體,廣泛地分布在中樞神經系統。
1.2 NR2 NR2是多基因家族,分別編碼為NR2A、NR2B、NR2C、NR2D。其中NR2A和NR2B對NMDA受體的結構和功能十分重要。含NR2A或NR2B的NMDA受體有突觸后密集區蛋白(PSD-95)等[1]一些相同的結合配偶體,NR2A可以與Homer蛋白、β-Catenin蛋白[2]和Rab親和蛋白3A結合[3],在成人腦部中主要表達在突觸內[4];而NR2B則與突觸RasGTP酶激活蛋白(SynGAP)等結合,在成人腦部中表達在突觸外[5],兩者較其他NMDA受體亞型都有較大的單通道電導系數,對胞外鎂離子的阻斷更敏感,鈣離子滲透率更大[6]。
NR2亞單位分布不同,且在成長過程中也會變化。在胚胎時期,NR2B和NR2D是主要的亞單位,前者表達于中樞神經系統中,后者只表達在間腦和腦干;出生兩周后,NR2A在中樞神經系統的表達逐漸增多,NR2B的表達在出生后7~10天達到高峰并限制在前腦區域,GluN2C出現較晚且限制在小腦和嗅球中,GluN2D的表達則在出生后發育而下降。NR2B與NR2A有著代償的聯系,減少突觸NR2B的表達可使NR2A的表達增加[7],且抑制因子-1沉默轉錄因子(REST)參與了NR2B向著NR2A隨年齡增大而成熟的轉變[8]。
1.3 NR3 NR3主要在發育中的中樞神經系統中表達,NR3經過不同的剪接得到兩個成員:NR3A和NR3B。NR3A在胚胎時期含量較低,但出生后很快升高,在青春期減少,主要分布于海馬、皮質和丘腦等;NR3B主要分布于腦干和脊髓的軀體運動神經元,NR3單獨不能形成功能受體,但是NR3可以與NR1和NR2形成NMDA受體復合物,起到負性調節的作用。
1.4 NMDA受體異四聚體的組成 功能性的NMDA受體是一個由兩個必需的NR1亞基和兩個NR2亞基或NR3亞基構成的異四聚體(見圖1),這些亞基結構高度相似,進而構成胞外的氨基末端域(amino-terminal domain, ATD)、胞外的配體結合域(ligand binding domain,LBD)、跨膜區(transmembrane domain,MD)和胞內羧基末端域(carboxy-terminal domain,CTD),與ATD相連的LBD進而與MD連接形成離子通道,MD的螺旋結構與CTD相連[9]。LBD由S1和S2兩個子結構域,其中在上部的S1結構具有一定剛性并與ATD相連,而在下部的S2結構具有一定的可變范圍并與MD相連,LBD與MD相連的可變性對于形成NMDA受體離子通道結構有著重要的作用。MD有3個跨膜的螺旋結構M1、M3和M4以及成孔凹環的M2, M3在谷氨酸門控型離子通道中有著最保守的片段,有關于與NMDA受體相似的AMPA受體的研究推測MD的打開是通過M3的自轉或遠離M2中心的側向位移而完成[10];M2尖端有一個關鍵的QRN位點決定了鈣離子對通道的滲透性。NR2A或NR2B的CTD有很多可以影響到NMDA受體活性的蛋白質相互作用和磷酸化位點,鼠海馬神經元NR2A的CTD中的羥基端與PSD-95的相互作用介導著NMDA受體的聚集分布[11]。
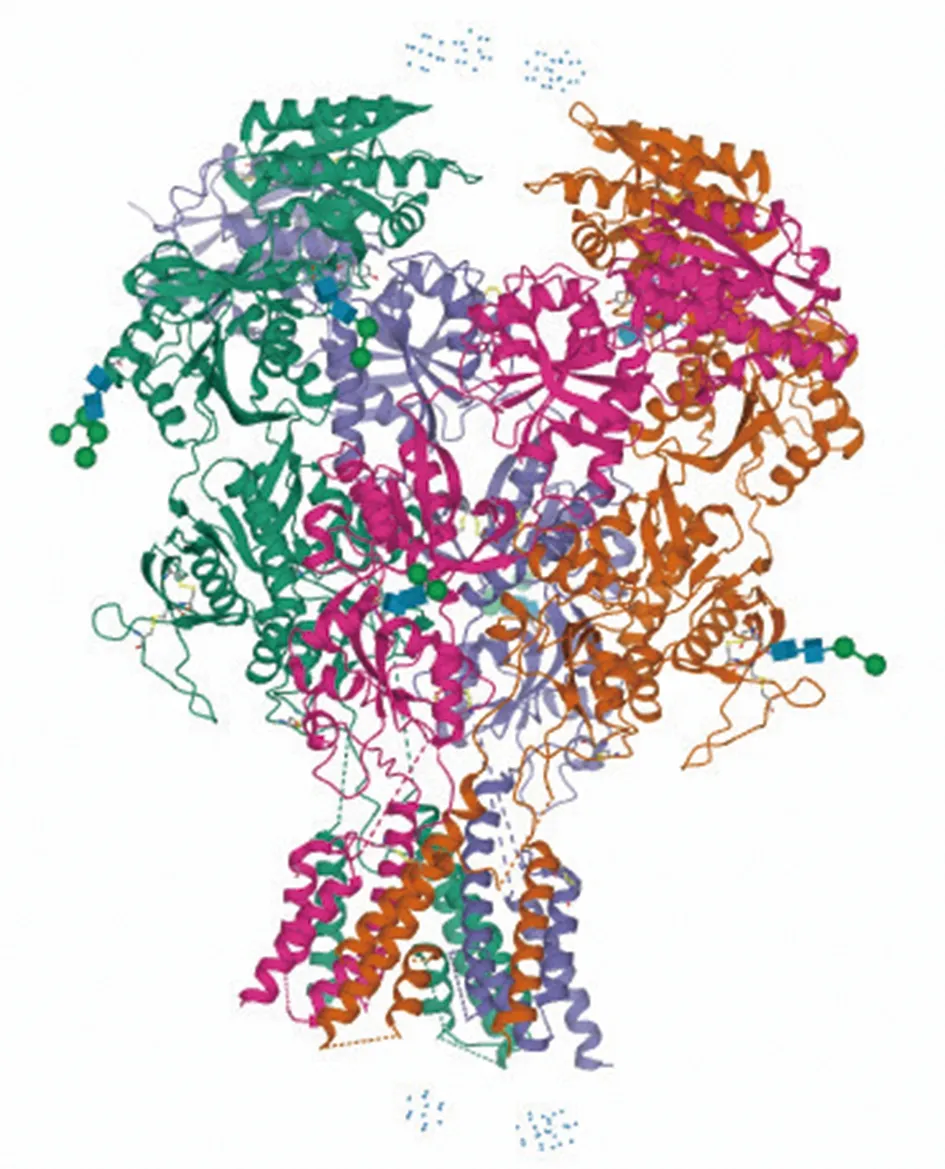
圖1 GluN1A/GluN2B NMDA 受體離子通道的晶體結構
可以看出,NMDA受體的結構和分布整體表現出的特點與其激活或抑制的狀態以及生理活性有關。NMDA受體結構上的不同結合位點、不同亞基組成的亞型不同在分布上有各自的特點,它們有各自的時空變化特點,存在獨特和交叉的部分,提示與個體生長發育過程各種生理功能的成熟有關。
2 NMDA受體的生理活性
亞基不同的NMDA受體激活后產生的生理活性有差異。
2.1 GluN1亞基的生理活性 GluN1亞基是所有NMDARs的重要組成部分,與NR2和/或NR3的兩個亞基組成NMDA受體通道。此外,GluN1亞基上存在甘氨酸 (Gly) 結合位點,調節NMDA受體的激活。GluN1亞基也與神經元細胞死亡有關,有文獻報道GluN1亞基氮-末端域(N-terminal domain,NTD)的一種配體,即組織型纖溶酶原激活劑(tissue-type plasminogen activator,tPA)調整著GluN1-NMDARs動力學從而控制著神經元的死亡[12], Castillo-Gómez等[13]研究發現針對GluN1-NMDARs的自身抗體存在致病潛力。
2.2 GluN2亞基的生理活性 NR2亞基分為NR2A、NR2B、NR2C和NR2D4四種。NR2A調節著神經元NMDA受體誘導的小神經膠質細胞與神經元細胞的物理相互作用[14],減少齒狀顆粒神經元中NR2A-NMDARs的表達,顯著抑制樹突生長[15],甘氨酸通過觸發NR2A-NMDARs非離子移變的活性而發揮了神經保護作用[16]。NR2B-NMDARs在神經遷移和皮質分層中扮演著不可缺少的角色,表達在谷氨酸能突觸的NR2B-NMDARs直接加速上升途徑突觸的細化[17],其活性的正反饋對于青少年學習過程的視覺記憶有著啟動的作用[18],在agouti相關肽神經元中參與了對體重平衡和血糖平衡的中央控制[19]。至于NR2C-NMDARs則在局部缺血后介導著神經保護作用[20],NR2C敲除模型小鼠表現出精神分裂癥樣的異常,如認知障礙和前脈沖抑制缺陷,在氯胺酮誘導的行為敏感性的維持上有重要作用[21],與NR2B-NMDARs一起促成丘腦底核中突觸的活性。另外突觸前包含NR2B、NR2C和NR2D的NMDA受體在孤束核可能控制著迷走神經的傳入興奮性。
2.3 GluN3亞基的生理活性 GluN3亞基有GluN3A和GluN3B兩種亞型。在亨廷頓氏舞蹈病動物模型中,GluN3A通過增強突觸傳導而促進NMDA生成[22],表達在嗅覺系統的GluN3A與嗅覺系統的發育有關[23]。NR3A在早期發育期間在CNS中廣泛表達[24],而NR3B在成人的運動神經元群體中富集[25]。
由此可見,亞型不同的NMDA受體的活性存在著交叉和差異,這是NMDA受體成為治療神經性疾病靶點的一部分困難所在。NMDA受體的過度激活會導致神經系統中突觸功能發生改變,進而引起中風、外傷性腦損傷、亨廷頓氏舞蹈病、阿爾茲海默病、精神分裂癥和抑郁癥等的發生,抑制NMDA受體的活性可以減輕興奮毒性,預防和減緩神經元的損傷。
3 NMDA受體拮抗劑
NMDA受體具有5個不同的結合位點,分別為①遞質結合位點 ;② 甘氨酸調節位點;③離子通道孔結合位點;④ 多胺調節位點;⑤ Zn2+結合位點。根據結合位點的不同,分為不同的靶向NMDA受體的藥物,下面主要介紹靶向NMDA受體甘氨酸位點、多胺調節位點及離子通道孔結合位點的藥物。
3.1 甘氨酸位點 甘氨酸在可以作為NMDA受體必不可少的共同配體,結合到NMDA受體甘氨酸結合位點上,促進NMDA受體的活性,甘氨酸也可以直接激活NR3-NMDA受體,具有興奮性遞質的功能。
3.1.1 L-701,324 L-701,324(2-氯-1-羥基-7-苯氧基苯基喹啉酮)是一種有效的NMDA拮抗劑,通過阻斷其甘氨酸B結合位點來拮抗NMDA受體的活性(結構式見圖2)。L-701,324可用于緩解焦慮、緊張和焦慮障礙、促進鎮靜,用于預防癲癇發作或降低其嚴重程度的藥物,L-701,324在小鼠中具有抗抑郁藥樣活性,部分是通過促進海馬BDNF系統介導的[26]。
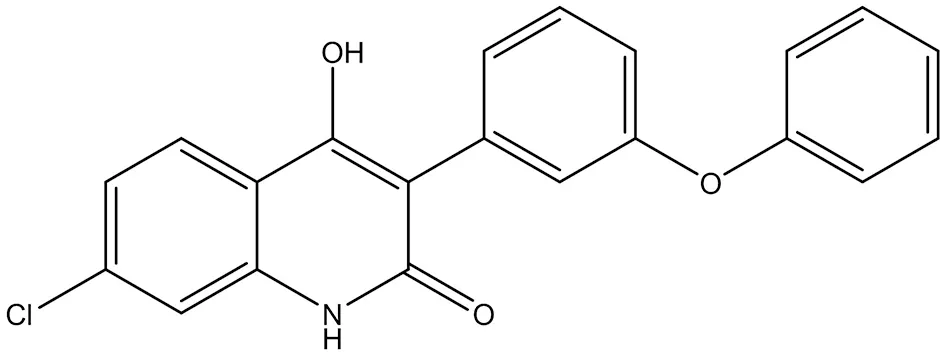
圖2 L-701,324結構式
3.1.2 ACEA-1021 ACEA-1021(6,7-二氯-5-硝基-1,4-二氫喹喔啉-2,3-二酮)與具有納米摩爾親和力的NMDA受體的甘氨酸位點結合,并且對非NMDA(AMPA / kainate)受體表現出相對較少的親和力[27](結構式見圖3)。在癲癇發作之前或之后立即用ACEA-1021治療后,可以防止高達86%接受致命劑量可卡因的小鼠死亡[28]。
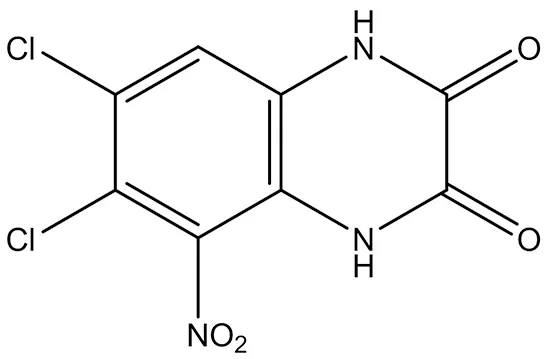
圖3 ACEA-1021結構式
3.1.3 GLYX-13(Rapastinel) GLYX-13 是一種N-甲基-D-天冬氨酸受體 (NMDAR) 甘氨酸位點功能部分激動劑和認知增強劑,也顯示出快速的抗抑郁活性,無精神分裂副作用(結構式見圖4)。用于治療重度抑郁癥(NCT03614156,NCT03560518),GLYX-13通過增強導水管周圍灰質中的AMPA受體功能來緩解慢性應激誘導的抑郁樣行為[29]。
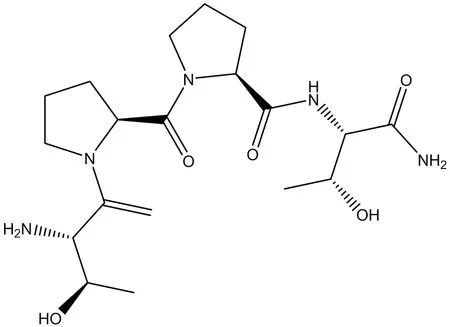
圖4 GLYX-13結構式
3.1.4 AV-101 AV-101(2-氨基-4-(2-氨基-4-氯苯基)-4-氧代丁酸) 是NMDA受體GlyB位點的選擇性拮抗劑(結構式見圖5),在雙盲隨機并有安慰劑對照的Ⅰ期臨床試驗中顯示出其安全性高和藥物動力學特點良好,可以用于治療神經性疼痛甚至是抑郁癥[30]。
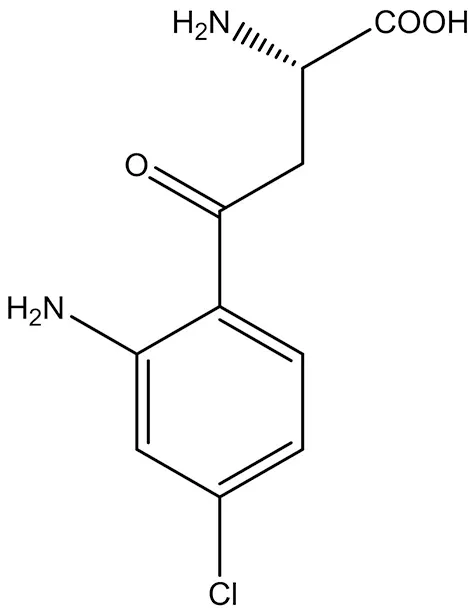
圖5 AV-101結構式
3.1.5 D-環絲氨酸(D-Cycloserine) D-環絲氨酸(4-氨基-1,2-惡唑烷-3-酮)是NMDA受體的共激動劑(結構式見圖6),在臨床上對精神分裂癥患者的神經可塑性沒有影響,在LTP測試中表現出很大的前高頻視覺刺激神經反應,說明D-環絲氨酸能結合NMDA受體[31],且在仍未結束的一個臨床試驗中被用于治療抑郁癥(NCT03062150)。
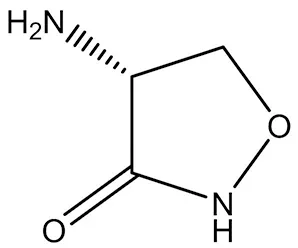
圖6 D-環絲氨酸結構式
3.2 多胺結合位點 廣譜的NMDA受體拮抗劑能影響所有NMDA受體而產生嚴重的精神副作用 ,限制了其臨床運用,因此,選擇性作用NR2B的NMDA受體拮抗劑成為更安全、有效的藥物。
3.2.1 MK-0657 (CERC-301) MK-0657 (4-甲基芐基(3S,4R)-3-氟-4‐[(嘧啶-2-ylamino)甲基]哌啶-1-羧酸酯)是一種口服生物可利用的選擇性N-甲基-D-天冬氨酸(NMDA)受體亞基2B(GluN2B)拮抗劑(結構式見圖7),目前正處于Ⅱ期臨床試驗中(NCT01941043,NCT02459236),其抗抑郁作用的工作機制尚不清楚,Lei等[32]研究發現MK-0657緩解了慢性約束應激(CRS)誘導的小鼠外側韁中的絕望樣行為,這種緩解可能涉及LHb中BDNF表達的降低,從而降低神經元活性。

圖7 MK-0657結構式
3.2.2 PEAQX PEAQX ([[[1S)-1-(4-溴苯基)乙基]氨基]-(2,3-二氧代-1,4-二氫奎噁啉-5-基)甲基]膦酸)是一種選擇性GluN2A拮抗劑(結構式見圖8),可用于治療皮質播散性抑郁癥[33]及精神分裂癥[34]。Mares等[35]的研究結果表明,GluN1/GluN2A首選拮抗劑PEAQX的抗驚厥作用具有年齡依賴性差異。
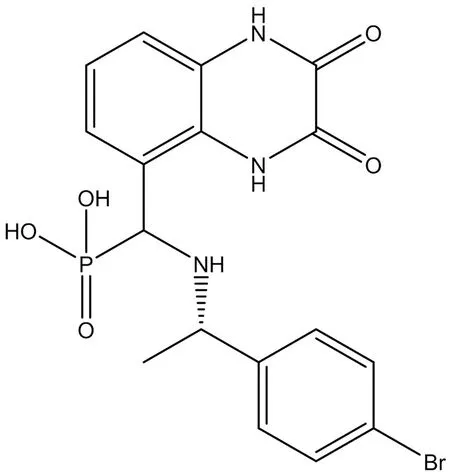
圖8 PEAQX結構式
3.2.3 艾芬地爾(ifenprodil) 艾芬地爾(4-[2-(4-芐基哌啶-1-基)-1-羥丙基]苯酚)是一種口服生物可利用的N-甲基-D-天冬氨酸(NMDA)受體拮抗劑(結構式見圖9),用作腦血管擴張劑[36],并在臨床試驗中用于治療藥物成癮[37],特發性肺纖維化和COVID-19。Ifenprodil結合并抑制谷氨酸NMDA受體GluN2B,從而防止NMDAR信號傳導。抑制了神經元的興奮性毒性,從而潛在地增強了認知功能。Ifenprodil可快速改善抑郁樣行為,激活mTOR信號傳導并調節CUMS大鼠海馬體中的促炎細胞因子[38]。一項關于ifenprodil治療COVID-19確診住院患者的安全性和有效性的研究正在進行2b/3期臨床試驗(NCT04382924)。
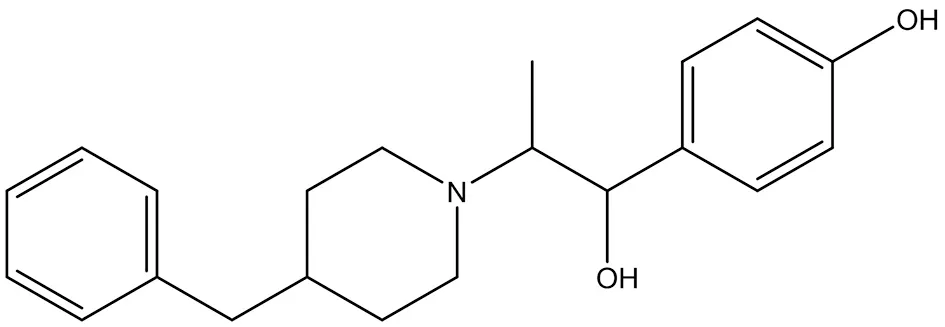
圖9 艾芬地爾結構式
3.3 離子通道孔位點 非競爭性NMDA受體拮抗劑能與NMDAR離子通道孔深部的PCP位點結合,阻斷與NMDAR耦聯的鈣通道, 減少Ca2+內流,抑制NMDAR的受體-通道的活動。目前發現的作用于NMDAR離子通道孔位點的藥物主要包括:苯環己哌啶 (phencyclidine, PCP) 、地卓西平 (dizocipine, MK-801) 、氯胺酮 (ketamine)、美金剛 (memantine) 、拉尼西明 (lanicemine, AZD6765) 、氧化亞氮 (nitrous oxide,N2O)。
3.3.1 Dizocipine(MK-801) MK-801是NMDA受體(受體,N-甲基-D-天冬氨酸)的強效非競爭性拮抗劑(結構式見圖10),影響認知功能、學習和記憶。它具有NMDA受體拮抗劑,麻醉劑,抗驚厥藥,煙堿拮抗劑和神經保護劑的作用。由于其嚴重的精神副作用,如幻覺、妄想、言語貧乏、意志減退等,禁用于臨床,其使用主要限于動物和組織實驗[39]。
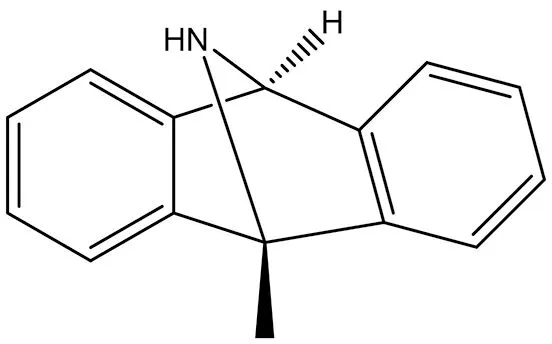
圖10 MK-801結構式
3.3.2 氯胺酮(ketamine) 在細胞實驗中,氯胺酮(結構式見圖11)通過抑制PKC/ERK通路而引起海馬神經元的凋亡可以被興奮性的NMDA受體激活所反轉[40]。氯胺酮表現出快速的降低抑郁癥患者情緒低沉程度的效果,這種效果可能是基于其改變額頂骨連接模式的能力[41],并且其代謝產物2R,6R-hydroxynorketamine對AMPA受體有興奮活性和抗抑郁的藥理活性,值得注意的是,這個代謝產物可能表現出更少的氯胺酮相關副作用[42];但是其對NMDA受體的抑制能力卻弱于氯胺酮,似乎2R,6R-hydroxynorketamine的抗抑郁作用不是完全由于其抑制NMDA受體的活性[43]。最近的一項實驗顯示,氯胺酮誘導催眠效果和神經可塑性是通過破壞磷酸化MAPK激酶(p-MEK)與磷酸化p-ERK的偶聯,下調p-ERK水平并上調磷酸化Fas相關死亡域蛋白(phosphorylated Fas-associated with death domain protein,p-FADD)水平[44]。氯胺酮作為經典的NMDA受體拮抗劑,曾經在臨床試驗中用于術后止痛(NCT02950233)[45]、重度抑郁癥(NCT03609190)[46],現在也有用于耳鳴(NCT03336398)、酒精復發(NCT02649231)和難治性抑郁癥(NCT02782104)的臨床試驗正在進行。
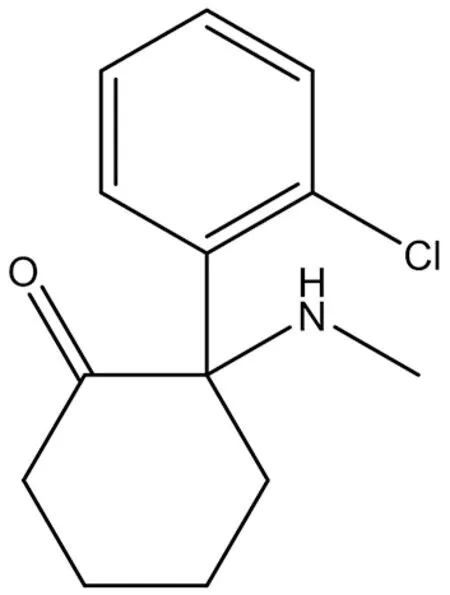
圖11 氯胺酮結構式
3.4 其他NMDA受體相關藥物
3.4.1 右美沙芬(dextromethorphan) 右美沙芬是非競爭性的NMDA受體拮抗劑(結構式見圖12),由Roche 公司開發,曾在臨床試驗中用于抑郁癥(NCT02860962,NCT02153502)和精神分裂癥(NCT02477670),現有正在進行的臨床試驗用于治療化療所致外周神經病變(NCT02271893)、亨廷頓病(NCT03854019)和癡呆型激動癥(NCT02446132),臨床上主要是用于鎮咳。研究顯示,右美沙芬對血管性癡呆(vascular dementia)大鼠的海馬神經損傷和認知能力缺陷有預防作用[47],但由于其與5-HT受體的作用可能導致5-羥色胺綜合征,會出現嘔吐、惡心、腹瀉和嗜睡等副作用[48]。

圖12 右美沙芬結構式
3.4.2 金剛烷胺(amantadine) 金剛烷胺(結構如圖13所示)也是非競爭性的NMDA受體拮抗劑,曾被用于治療PD、藥物導致的錐體束外反應以及病毒感染病等,在人體中可能導致反副交感神經生理樣副作用(如口干、尿潴留、便秘、惡心、頭暈和失眠等)。最近的研究顯示[49],金剛烷胺增強大鼠運動和探尋活動相關的黑質紋狀體和中腦緣的多巴胺功能,在一項隨機雙盲試驗中[50],Nourbakhsh等[51]發現金剛烷胺改善多發性硬化疲勞方面并不優于安慰劑,并導致更頻繁的不良事件。
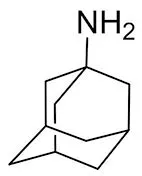
圖13 金剛烷胺結構式
3.4.3 石杉堿A(huperzine A) 石杉堿A(結構如圖14所示)為蛇足石杉(Huperziaserrata)中成分,已批準用于治療AD,是一個選擇性的AChE拮抗劑和非選擇性的NMDA受體拮抗劑,有抗炎、鎮痛和抗痙攣作用[52],陳慶狀等[53]研究發現HupA可通過減少Aβ與淀粉樣蛋白結合醇脫氫酶(ABAD)的結合而改善線粒體損傷,進而改善AD小鼠的認知和記憶功能障礙。曾在臨床試驗中用于精神分裂癥(NCT00963846)和癡呆癥(NCT01012830),也有用于外傷性腦損傷(NCT01676311)和提高認知能力(NCT01676311)的臨床試驗正在進行,另外一項實驗證明飲食誘導的肥胖小鼠中,Hup A治療可以有效地改善認知功能[54]。
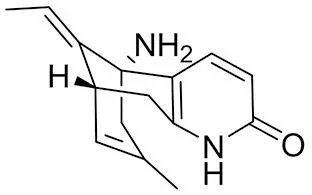
圖14 石杉堿A結構式
現將與NMDA受體有關的藥物總結如表1所示。
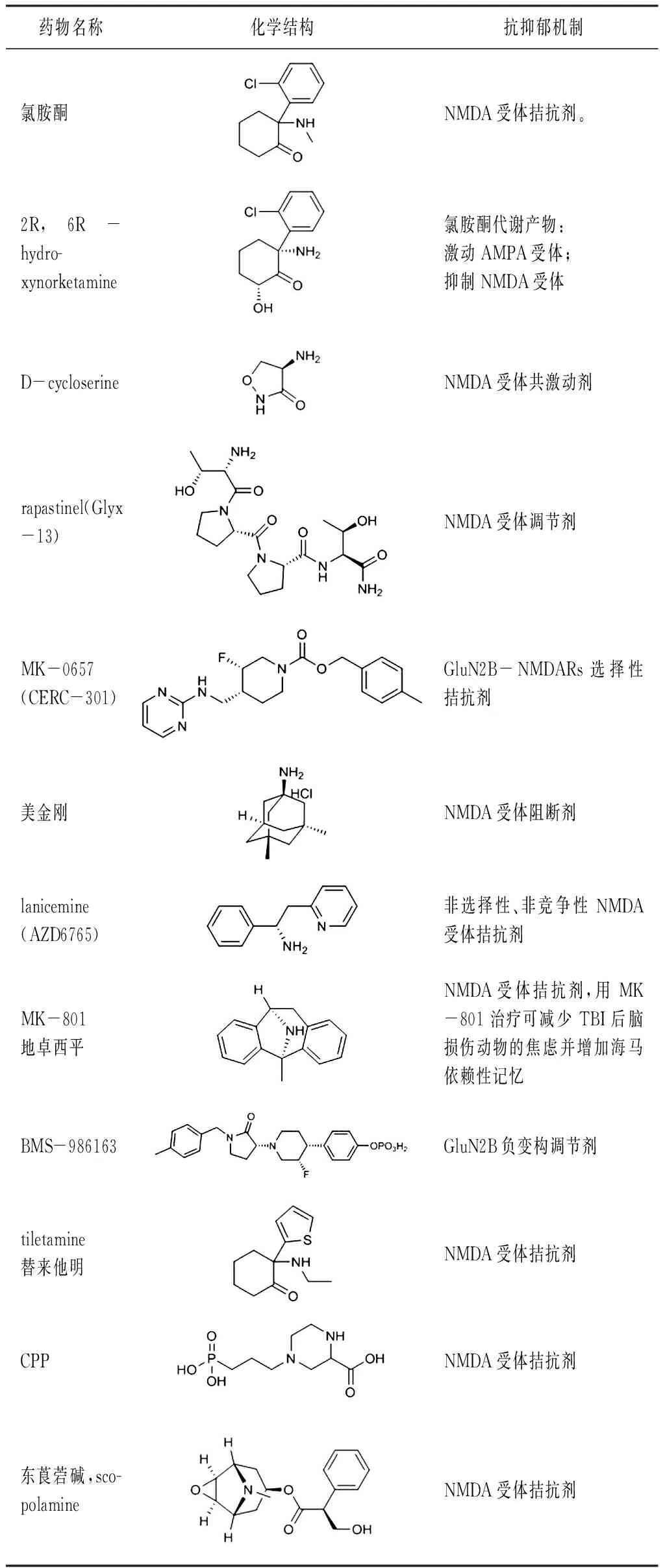
表1 NMDA受體相關藥物
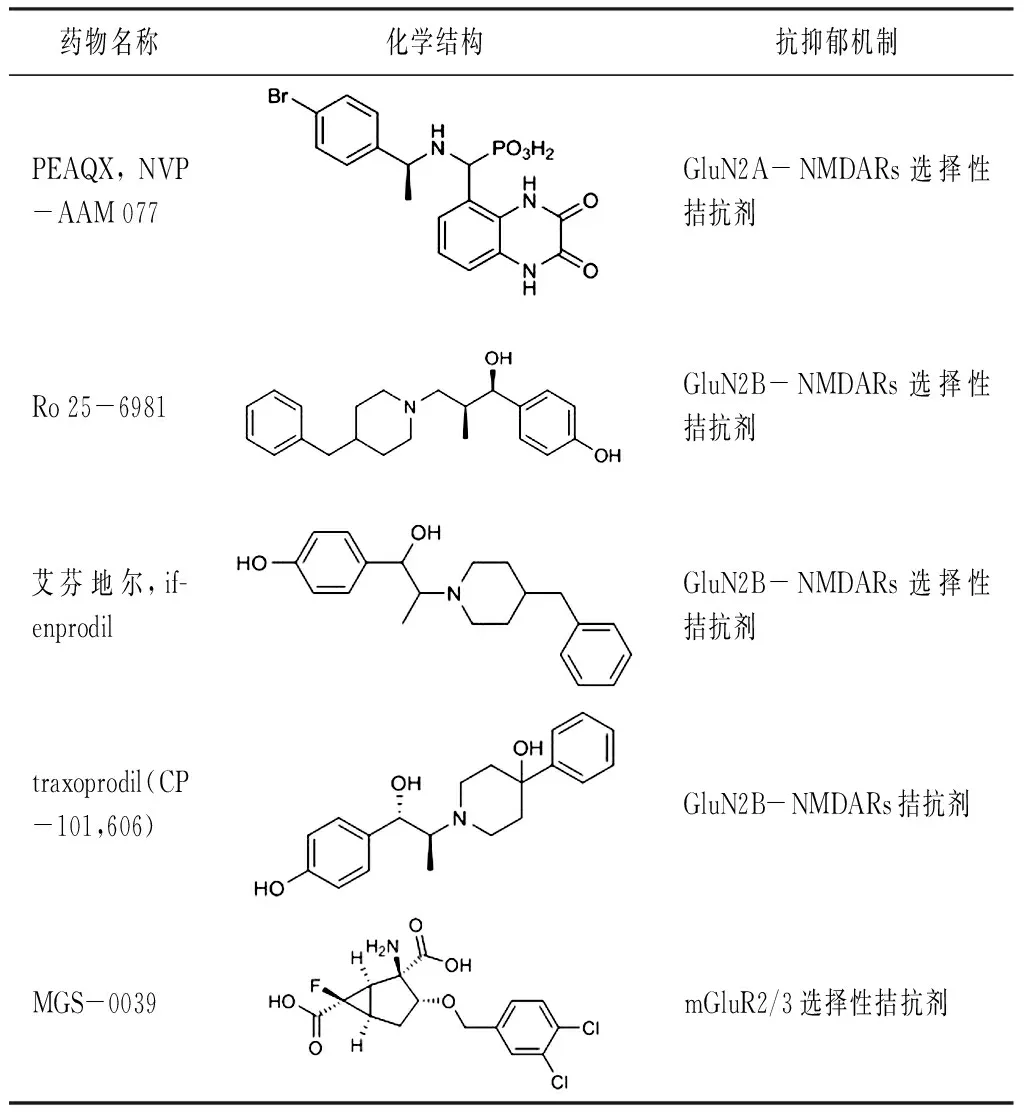
表1(續)
4 總結與展望
至此,NMDA受體在神經性疾病中的重要位置已不言而喻,其作為治療和預防神經性疾病的靶點的潛力大,但由于分型的高度同源和其廣泛的生理活性,單一化合物針對性成藥較難,溫和的、亞型選擇性強的NMDA受體調節劑又或聯用共激動劑和變構調節劑有著更好的成藥前景,由回顧前人的研究成果與已經上市的有關NMDA受體的藥物可見。因此,接下來的研究方向便可能為:①已有NMDA受體拮抗劑的結構改造,以增加其對不同位置、不同亞型的NMDA受體的選擇性;②尋找更具選擇性的變構調節劑或共激動劑;③通過基礎研究構建與谷氨酸能神經生理活動相關的生理信號系統,通過間接的靶向其他重要靶點以影響NMDA受體和有關的生理信號通路,以達到調節谷氨酸能神經功能的目的;④進一步研究藥物相互作用,尋找在治療作用和副作用上互補或協同的藥物組合,以達到系統地調節NMDA受體活性的目的。
我們有理由相信,隨著更多的研究成果的浮出,關于NMDA受體的探索將會在神經領域中繼續深入,治療和預防神經性疾病的研究道路由此開辟。
