內相粒徑對現場混裝乳化炸藥非等溫熱分解特性的影響
2022-05-05 13:23:38吳攀宇李松林陳皓楠
火炸藥學報 2022年2期
關鍵詞:質量
吳攀宇,劉 鋒,魏 國,李松林,何 祥,陳皓楠
(安徽理工大學 化學工程學院,安徽 淮南 232001)
引 言
近年來,隨著國民經濟與爆破技術的發展,炸藥需求用量不斷增大,現場混裝乳化炸藥技術憑借其高度機械化與自動化的優勢,成為行業未來發展的主要方向[1]。由于乳化炸藥及其基質的熱力學亞穩態體系在使用中發生多起燃燒、爆炸事故[2-3],其熱穩定性研究受到廣泛重視。朱帥等[4]使用TG技術研究了復合油相對乳化炸藥熱分解特性的影響;尹利等[5]使用DSC技術研究了不同敏化方式對巖石型乳化炸藥熱分解過程的影響;馬志鋼等[6]使用DSC-TG聯用技術研究了含水量對乳化炸藥基質熱分解特性的影響,發現含水量高的乳化炸藥基質放熱速度和質量損失速度更快;李洪偉、羅寧等[7-8]使用C80等技術研究了復合乳化劑對乳化炸藥熱分解行為的影響,發現使用復合乳化劑制備的乳化炸藥穩定性均高于使用單一乳化劑的乳化炸藥;劉鋒、吳攀宇等[9-10]使用TG-DSC聯用技術研究了不同氣氛與外界燃料油對乳化炸藥熱分解特性的影響。以上研究著重分析了油相、水相、乳化劑、外界氣氛與工況等對乳化炸藥或其基質熱分解的影響,但未見現場混裝乳化炸藥內相粒徑對其熱分解特性影響等方面的研究報道。
作為第二代露天乳化炸藥現場混裝技術的核心參數,乳化炸藥基質的內相粒徑決定了其微觀結構和在儲運、泵送和使用中的穩定性[11-12]。由文獻[11]可知,由于乳化炸藥的內相沸點比外相更高,受熱時外相先于內相汽化,所以內相粒子的直徑大小影響到外相油膜厚度與對外界能量的吸收,進而影響乳化炸藥的熱穩定性、感度和爆速等宏觀性能。并且,在乳化炸藥的制備過程中,轉速越高,內相粒徑越小,黏度就越大,不利于現場裝填,且兩相之間的劇烈撞擊運動易加速形成“熱點”,生產安全性低;制備轉速越低,內相粒徑越大,粒徑分布越寬,乳化炸藥的穩定性與爆炸性能隨之變差[1]。因此,為確保現場混裝乳化炸藥的安全生產與穩定使用,有必要研究內相粒徑對其基質熱分解特性的影響。
本研究采用激光粒度儀和光學顯微鏡對現場混裝乳化炸藥基質進行了粒徑測試和微觀結構觀察,利用TG-DTG技術測試了不同粒徑基質的非等溫熱分解過程,計算了熱分解動力學參數,推導了熱分解動力學機理函數,以期為現場混裝乳化炸藥的安全生產與穩定使用提供指導。
1 實 驗
1.1 試劑與儀器
硝酸銨(AN)、硝酸鈉(SN),工業級,國藥集團化學試劑有限公司;復合油相,工業級,河北國控化工集團有限公司;Span-80,AR,阿拉丁試劑(上海)有限公司;0#柴油。
Malvern Mastersizer 2000激光粒度儀,馬爾文帕納科公司;XSP-86系列無限遠生物顯微鏡,上海田瞳光學科技公司;DSCQ2000型TG差示掃描量熱分析儀,梅特勒-托利多公司;JFS-550型變頻多用分散器,杭州齊威儀器有限公司。
1.2 現場混裝乳化炸藥基質的制備
水相和油相的制備:按照表1配方,將稱量好的硝酸銨、硝酸鈉和水混合加熱至90~100℃;將稱量好的復合蠟、柴油、Span-80加熱至80~90℃。

表1 現場混裝乳化炸藥基質的配方
制備過程與工藝:設定乳化炸藥基質的制備轉速分別為600、800、1000、1200、1400r/min,對應的樣品編號分別為樣品1、2、3、4、5。將分散器的轉速調至設定值,在40s內將水相溶液勻速加入到油相中,繼續保持均勻高速剪切3min制得乳化炸藥基質。純硝酸銨的對照樣品設為樣品6。
1.3 乳化炸藥基質微觀結構觀測
采用光學顯微鏡觀察乳化炸藥基質內相液滴外觀。制備觀測樣品的方法為:將蓋玻片和載玻片浸泡在質量分數5%鹽酸的酒精溶液中2h去污,用玻璃棒蘸取少量待測樣品放在干凈載玻片上,用毛細管滴加柴油稀釋至約10倍,攪動玻璃棒至均勻分散后蓋好蓋玻片放到顯微鏡下觀測,放大倍數為400倍。
1.4 乳化炸藥基質粒徑的測試
采用激光粒度儀測試乳化炸藥基質內相粒徑。測試方法與參數為:取5g乳化炸藥基質,使用玻璃棒手動分散于100mL柴油中,將質量分數控制在0.05%~0.15%,使用膠頭滴管將待測樣品均勻注入樣品池中,測試其表面積體積平均直徑D[3,2],每個樣品測兩次取平均值。
1.5 乳化炸藥基質的熱分析
采用TG-DTG技術測試乳化炸藥基質熱分解過程。測試參數為:樣品質量為(2±0.2)mg;分別以5、10、15、20K/min的升溫速率(β)由20℃升溫至400℃,使用Al2O3坩堝與N2氣氛,氣氛流速為100mL/min,系統自動采集數據。
2 結果與討論
2.1 樣品的微觀結構
樣品1~樣品6光學顯微鏡觀測結果如圖1所示。

圖1 樣品1~樣品6的微觀結構
由圖1可知,樣品1和樣品2中出現棱塊狀硝酸銨晶體,內相粒徑較大;樣品3~樣品5未見明顯的硝酸銨結晶。乳化炸藥的內相粒徑隨著制備轉速的提高而減小。自由能差異導致的吸收現象在樣品1和樣品2中尤為明顯,大液滴周圍存在大量小液滴[13]。
2.2 粒徑測試結果分析
樣品1~樣品5的粒徑測試結果見表2;樣品1~樣品5的內相粒徑分布見圖2。
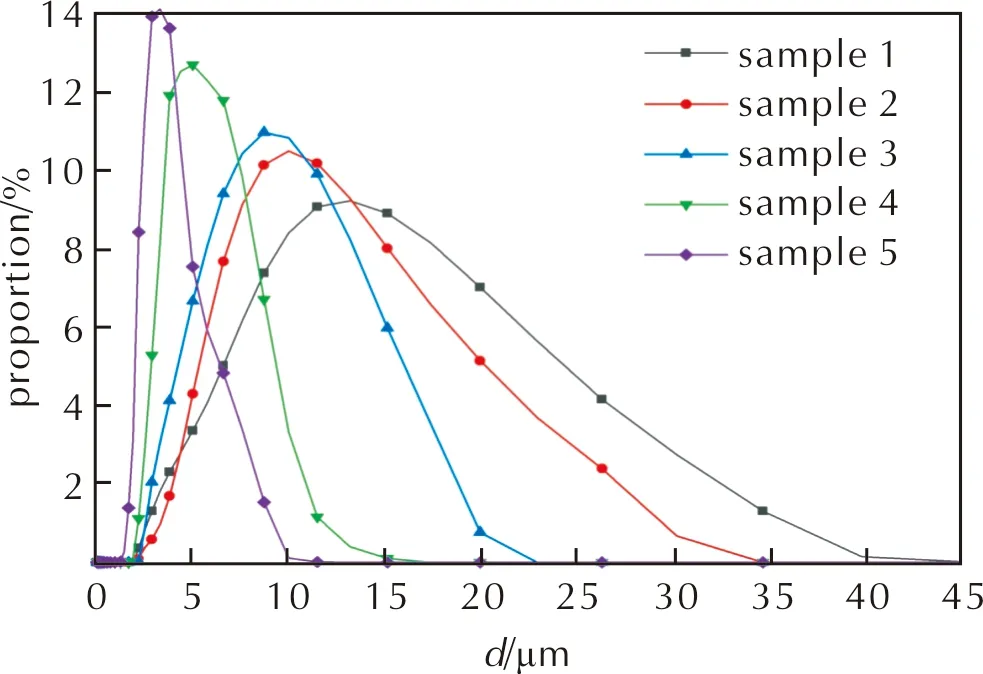
圖2 樣品1~樣品5的粒徑分布圖
結合圖1、圖2與表2可知,激光粒度儀的粒徑測試結果與光學顯微鏡的觀察結果吻合。根據文獻[14],對于高內相的乳狀液,可通過計算粒徑分布范圍與平均粒徑的比值表征其多分散指數(PDI),計算結果見表2。
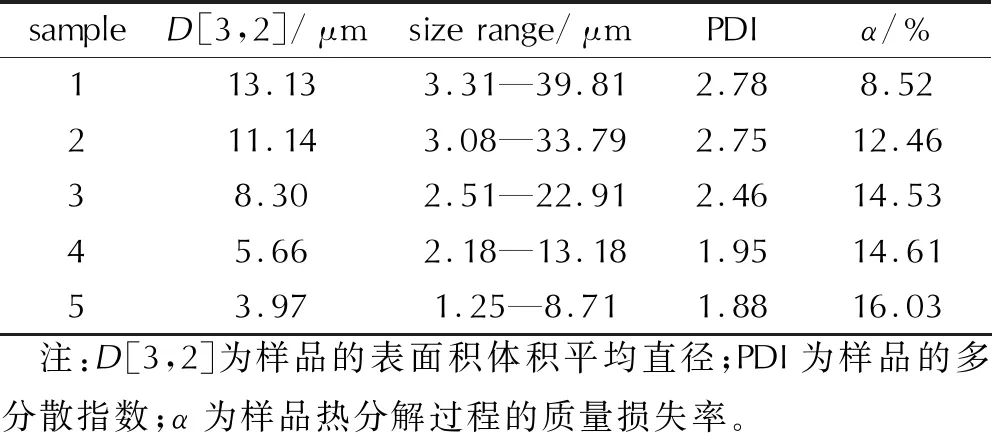
表2 樣品1~樣品5的粒徑測試結果
分析認為,乳化炸藥基質的制備轉速不同時,分散機剪切速率不同。低轉速制備的乳化炸藥基質受到的剪切應力較小,內相粒徑更大,發生奧氏熟化與聚合等過程的概率與程度增大[1,15],樣品1和樣品2的內相D[3,2]大于10μm,粒徑分布在3.08~39.81μm的較大區間,PDI較大,體系均一性較差,穩定性較差,易從液滴內部發生析晶而導致性能下降[1,14]。熟化或聚合后內相液滴直徑差異增大,導致粒子自由能差異增大,具有較低自由能的大粒子聚合吸收小粒子的趨勢增加,加速大粒子不斷增大、小粒子不斷縮小的過程,系統總自由能降低[13,15]。對于樣品4和樣品5,乳化器保持高速剪切,D[3,2]被控制在5μm左右,基質內部發生熟化與聚合現象的程度降低,粒徑分布范圍為1.25~13.18μm的較小區間,PDI減小,粒徑分布集中,均一性好,基質內部未見熟化、吸收等導致的明顯粒徑差異,整體保持較高的穩定性。
2.3 熱分解過程分析
乳化炸藥基質樣品與純AN的TG曲線見圖3,DTG曲線見圖4,其中代表較大粒徑是樣品1,代表較小粒徑的是樣品4。
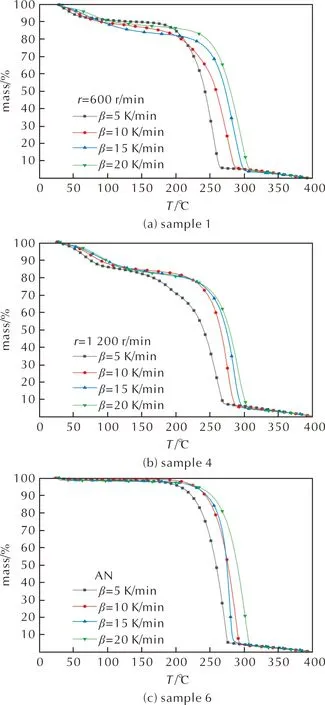
圖3 樣品1、樣品4和樣品6的TG曲線
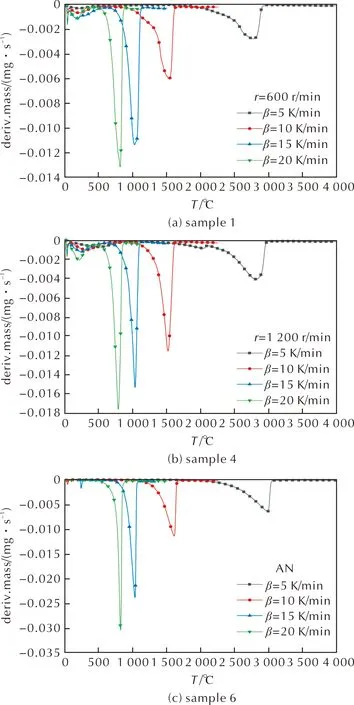
圖4 樣品1、樣品4和樣品6的DTG曲線
結合圖3和圖4可知,在線性升溫條件下,乳化炸藥基質的受熱分解存在兩個典型的質量損失過程,分為3個階段。第一階段在TG曲線上表現為在20~110℃,質量損失率(α)約為8%~16%,對應在DTG曲線上0~250s內出現明顯的質量損失速率峰,表明在第一階段乳化炸藥基質中的硝酸銨和燃料油相未發生顯著分解,可能是由于基質中的游離水受熱蒸發,脫離內相所導致[16]。該階段的質量損失程度隨著粒徑的減小而增大,不同粒徑乳化炸藥基質樣品在第一階段的質量損失率見表2,可能為樣品1和樣品2在制備后逐漸析晶,一部分水(主要是游離水)在制備(乳化溫度可達100℃[16])時已經脫離基質并蒸發所導致,粒徑較大的樣品乳化不完全,制備時失水較多,因此TG實驗中檢測出的水分較少,第一階段質量損失率較小。樣品5粒徑最小,均一性最好,做TG實驗前未出現兩相分離等失穩現象,TG實驗中檢測出的游離水含量更高。
第二階段對應溫度在110~170℃,由圖3可知,在此階段中樣品質量變化緩慢,無明顯質量損失。由圖4可知,對于同組樣品,升溫速率越快,該階段歷時越短,6組樣品在此階段無明顯的差異。
第三階段對應溫度在170~310℃,TG曲線驟降,表明反應物質量快速損失,發生劇烈的分解反應,α約為75%~80%,各組TG與DTG曲線隨著升溫速率的增大,向高溫方向有不同程度偏移。6個樣品的DTG峰溫(Tp)見表3,樣品在升溫速率10K/min下的反應起始溫度(Tonset)與劇烈分解階段的質量損失平均速率(v)見表4。
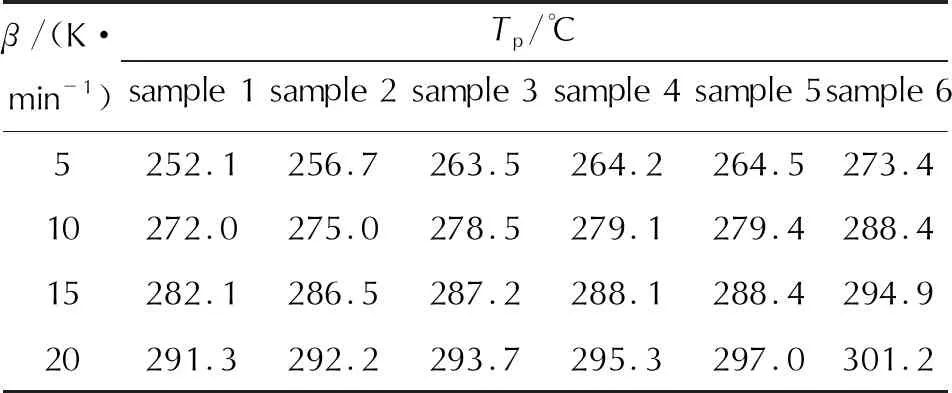
表3 6個樣品的DTG峰溫
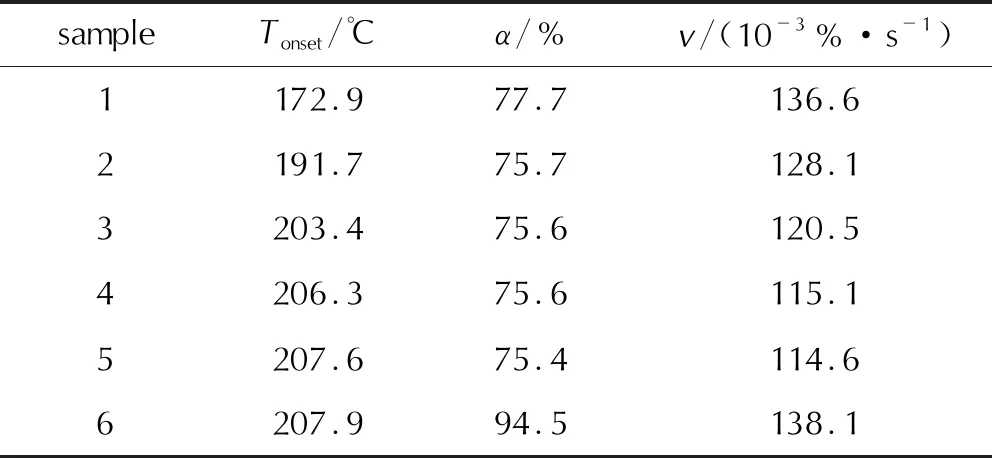
表4 6個樣品的反應起始溫度與質量損失平均速率
結合表3與表2可知,樣品1~樣品5的Tp隨著粒徑的減小而增大,整體呈現略向高溫端的移動。結合表4與表2可知,樣品1~樣品5的Tonset隨著粒徑的減小而不斷升高,說明乳化炸藥基質的熱穩定性隨著乳化炸藥基質內相粒徑的減小而增大。樣品6的分解過程質量損失率最大,質量損失平均速率最大;樣品1~樣品5的質量損失率受第一階段水分蒸發所影響,呈現減小趨勢,質量損失平均速率隨著粒徑的減小而減小,由于樣品1~樣品5是不均勻的油水混合物,其平均速率均小于樣品6純AN的。
2.4 動力學參數計算
樣品1~樣品5的熱分解起始溫度約為170~210℃,升溫速率為5K/min時對應的質量損失率為20%。根據圖1、圖2的TG-DTG測試結果,在α為20%~95%的范圍內,使用熱分析曲線動力學分析微分法(Kissinger法)計算動力學相關參數。Kissinger法方程為[17-18]:
(1)
式中:β為升溫速率,K/min;Tp為DTG峰溫,K;A為指前因子,s-1;Ek為反應活化能,kJ/mol;R為普適氣體常數,8.314J/(mol·K);
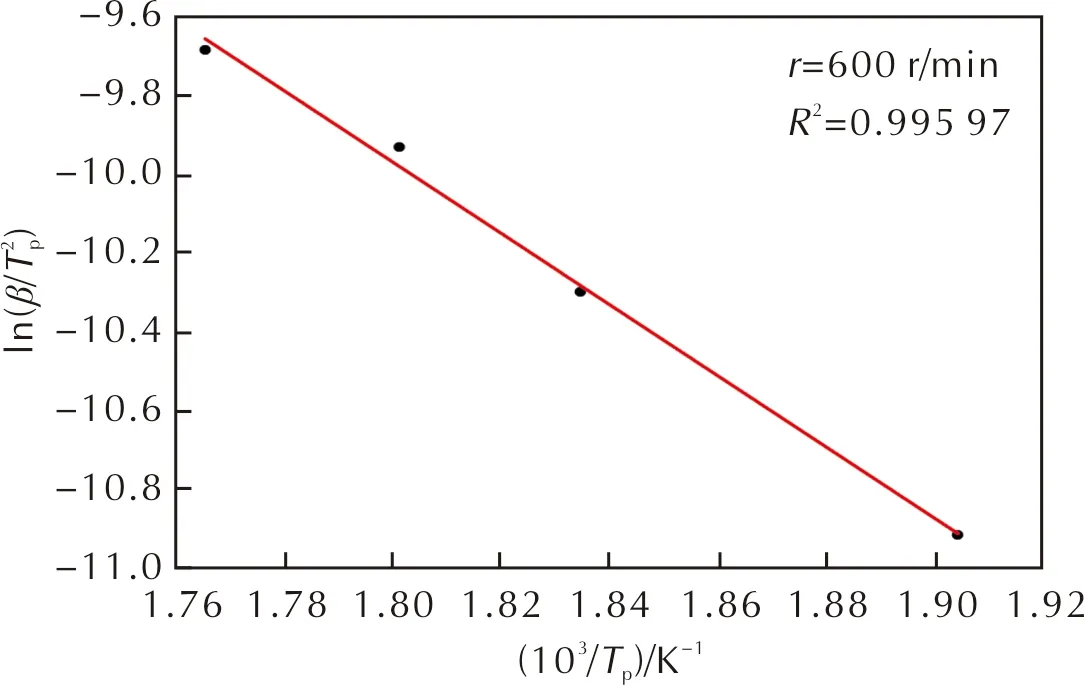
圖5 樣品1的圖
使用Kissinger法可計算樣品在α為20%~95%階段的動力學參數。對于乳化炸藥基質,其熱分解活化能隨著內相粒徑的減小而增大,粒徑大于10μm乳化炸藥基質的熱分解活化能約為80kJ/mol,粒徑小于5μm的基質熱分解活化能約為110kJ/mol,上升了37.5%。
2.5 動力學機理函數模型
將不同升溫速率的TG曲線上質量損失率(α)及其對應的溫度(T)代入Coast-Redfern法的48種動力學機理函數,尋求最概然動力學機理函數G(α)。Coast-Redfern方程為[17,19]:
(2)
式中:各參數物理意義同式(1)。
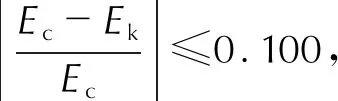

表5 擬合機理函數模型
由表5可知,各組實驗的最概然動力學機理函數存在差異:粒徑大于10μm乳化炸藥基質樣品(樣品1和樣品2)和純硝酸銨(樣品6)服從Valensi方程,屬二維擴散的圓柱形對稱模型;樣品3服從Jander方程,屬三維擴散球形對稱模型;粒徑約為5μm的樣品(樣品4和樣品5)服從Zhuralev-Lesokin-Tempelman方程,即Z-L-T方程,屬三維擴散控制模型。6個樣品活化能誤差值均在要求范圍內。乳化炸藥基質樣品的線性相關系數較差,分析認為乳化炸藥基質是混合物,當剪切力較低時,基質混合程度和均一性較高轉速制備的基質或純硝酸銨更差,熱分解過程更加復雜,其活化能隨著反應的進行而不斷變化,導致其線性相關系數較純硝酸銨更差。
2.6 內相粒徑與熱分解特性關系
2.6.1 內相粒徑與質量損失平均速率的關系
結合表2與表4可知,現場混裝乳化炸藥基質在第三分解階段的質量損失平均速率隨著內相粒徑的減小而減小;反應起始溫度隨著粒徑的減小而增大。
由文獻[1,11]可知,對于確定配方與制備工藝的乳化炸藥基質,當制備轉速較高時,內相粒子直徑小且分布均勻,液滴的比表面積大,連續相油膜厚度小,乳化劑分子密度低,液滴油膜傳熱效率增大,樣品受熱能夠均勻、快速地傳導,內相液滴汽化時間一致,樣品整體能夠同時達到最大“能量”;相反,當制備轉速較低時,內相粒子直徑較大,連續相油膜厚度增大,且制備時失水較多,基質整體水含量降低,樣品呈現局部溫度升高,粒徑小的粒子先反應,整體不會同時達到最大“能量”。內相粒徑較大的樣品,其粒徑分布范圍更廣,其中粒徑小的粒子首先急劇汽化,對外相有著強烈的爆裂作用,使得外相材料成為極細小的油滴懸浮在氧化劑分解后的產物中,繼續形成“熱點”,使得反應更加迅速;隨著乳化炸藥內相粒子直徑的減小、體系均一度的提高、反應起始溫度的上升,其內相粒徑達到整體汽化所需能量的時間明顯要長于分布更寬的大粒徑所需時間,反應速率系數會降低,表現為第三分解階段質量損失平均速率的降低或反應歷程時間的增大,符合實驗所得結果。
2.6.2 內相粒徑與動力學機理函數的關系
結合表2與表5可知,現場混裝乳化炸藥基質的動力學機理函數隨著粒徑的減小,由二維擴散變化為三維擴散動力學反應機理。分析認為,在170~310℃的第三分解反應階段,樣品1和樣品2以部分油水混合物狀態開始反應,所有參與反應粒子的成核幾乎同時發生,并且是一個瞬時隨機的過程,粒子表面或邊緣產生成核界面,隨后發生快速的二維生長,對于顆粒狀的樣品6,其動力學反應機理與樣品1和樣品2相同,均為二維擴散理論[20]。樣品4和樣品5中的硝酸銨與燃料油在第三分解反應階段前暫未發生顯著的破壞,油水相結構較為完整且均一,致密的粒子排布與更少的爆裂作用減緩了體系的熱積累和“熱點”的形成,從而降低了反應速率,進而延長了成長核的形成時間,大部分的成核位點重合,在轉為活躍狀態之前已經同成長核結合,導致其形成一個復雜形狀的擴散體,對應于三維成長,動力學反應機理屬三維擴散[20]。
2.6.3 內相粒徑與熱分解活化能的關系
由表2與活化能計算結果(Ek)可知,乳化炸藥基質的熱分解活化能隨著粒徑的減小而增大。分析認為,乳膠基質作為一種高內相比的乳狀液,其內相主要成分為硝酸銨的過飽和溶液,可對溶液應用開爾文定律研究[21]。
(3)
式中:α0為普通晶體在熱力學溫度T時刻的活度,mol/L;αr為微晶在T時的活度,mol/L;σ為晶體物質的表面張力,N/m;ρ為晶體物質的密度;M為晶體物質的摩爾質量;r為微晶的半徑。
由式(3)可知,當內相粒徑較大時,粒子內相溶液中可能發生硝酸銨的析晶,硝酸銨晶體易刺破表面活性劑膜,在液滴的毗鄰處發生析晶失穩[22-23]。
當內相粒徑逐漸減小時(樣品4和樣品5),基質的粒子均一性、整體穩定性不斷提升,油膜厚度的降低提高了體系的一致性,從而減緩了基質內部的熱累積速度,此外,爆裂作用的發生與“熱點”形成的難度增大、概率降低,延長了成長核從生成到活躍的時間,增大了反應物的反應能量位壘,宏觀上表現為乳化炸藥基質體系熱分解活化能的升高。
3 結 論
(1)隨著現場混裝乳化炸藥基質內相粒徑由13.13μm減小至3.97μm,其失水過程質量損失率由8.52%升高至16.03%,起始分解溫度由172.9℃升高至207.6℃,平均DTG峰溫由274.4℃升高至282.3℃,質量損失平均速率由0.136%/s降低至0.114%/s,熱穩定性不斷上升。
(2)使用Kissinger法計算了不同粒徑的現場混裝乳化炸藥基質在質量損失率α為20%~95%范圍的動力學參數。粒徑大于10μm的乳化炸藥基質的熱分解活化能約為80kJ/mol,粒徑小于5μm的基質熱分解活化能約為110kJ/mol,上升了37.5%。
(3)使用Coast-Redfern法擬合了不同粒徑的現場混裝乳化炸藥基質的最概然動力學機理函數。粒徑大于10μm的乳化炸藥基質的動力學反應機理受二維Valensi方程控制,粒徑小于5μm的基質動力學反應機理受三維Z-L-T方程控制。
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
