超聲波處理對兔毛角蛋白組成與結構的影響
2022-04-24 03:00:00王曉清史志銘李曉宇
紡織學報 2022年4期
關鍵詞:結構
王曉清, 史志銘, 李曉宇
(1. 內蒙古工業大學 輕工與紡織學院, 內蒙古 呼和浩特 010080; 2. 內蒙古工業大學 材料科學與工程學院, 內蒙古 呼和浩特 010051)
兔毛作為典型的特種動物纖維,資源豐富,價格低廉,是一種由角蛋白、微量色素以及灰分等組成的蛋白質纖維,其中角蛋白含量高達90%以上[1]。角蛋白由多種氨基酸通過二硫鍵、氫鍵、離子鍵和疏水作用連接形成四級結構[2],使兔毛具有穩定的化學性能和較強的力學性能[3]。然而由于特殊的髓質層和鱗片層結構,導致兔毛可紡性能較差,每年在紡紗過程中都會產生大量兔毛廢料,造成嚴重的資源浪費和環境污染,因此,將兔毛廢料轉化為有用的角蛋白材料引起越來越多的關注。
角蛋白具有生物相容性和生物可降解性,此外提取的角蛋白能夠形成調節細胞識別和行為的自組裝結構,因此,被認為是一種潛在的生物材料新資源,可用于組織醫學領域[4-5]。鑒于以上優點,且動物毛發資源豐富,近年來許多研究采用化學水解、氧化、還原等方法提取角蛋白[6-8],還有采用蒸汽閃爆輔助堿液以及離子液體提取角蛋白的報道[9-10]。角蛋白提取的原理主要是破壞纖維中胱氨酸的二硫鍵(S—S)形成半胱氨酸(—SH),使蛋白質大分子鏈斷裂,進而破壞角蛋白的各級結構,最終使纖維解體。通常采用增加化學試劑用量或提高溫度使二硫鍵的破壞程度增加,進而提高角蛋白的提取效率,但這會導致大分子主鏈上更多的肽鍵水解生成大量小分子多肽,在一定程度上限制了角蛋白的再次利用。
兔毛髓質層發達,結構均勻,分級排列,這種結構有利于超聲波空化作用[11],因此,本文采用超聲波輔助還原法對兔毛角蛋白進行提取,在一定程度上通過增加物理作用,降低化學作用對酰胺鍵的破環,在保證角蛋白提取效率的基礎上制得分子質量高的角蛋白,對提取的角蛋白結構進行表征,研究超聲波處理對兔毛角蛋白組成和結構的影響。
1 實驗部分
1.1 實驗材料和儀器
材料:兔毛,采自中國甘肅天水兔子養殖場,品種為德系安哥拉長毛兔;尿素,天津大茂化學試劑有限公司;亞硫酸氫鈉,天津化學試劑有限公司;十二烷基硫酸鈉(SDS),山東優索化工科技有限公司;石油醚、無水乙醇、氫氧化鈉、氫氧化鋰和鹽酸,國藥集團化學試劑有限公司;丙烯酰胺、N,N′-亞甲基雙丙烯酰胺、過硫酸銨、甘油、無水甲醇、冰乙酸、溴酚藍、甘氨酸(Gly),天津市福辰化學試劑有限公司;5,5′-二硫代-雙-(2-硝基苯甲酸)(DTNB)、三羥甲基氨基甲烷(Tris),恒遠博泰生物科技有限公司;考馬斯亮藍R-250,Sigma-Aldrich公司;四甲基乙二胺(TEMED)、 β-巰基乙醇、乙酸鈉緩沖液和Tris-HCl緩沖液,上海麥克林生化科技有限公司;乙二胺四乙酸(EDTA),天津市化學試劑一廠;截留分子質量為8~14 ku的透析袋,北京索萊寶科技有限公司。
儀器:LGJ-12A冷凍干燥機,北京四環起航科技有限公司;L-8900氨基酸分析儀,日本日立公司;LC-20AT高效液相色譜儀、UV-2700紫外-可見分光光度計、IR Affinity-1傅里葉紅外光譜儀,日本島津公司;DYCZ-24DN電泳儀,北京六一生物科技有限公司;inVia Reflex拉曼光譜儀,英國雷尼紹公司;D/MAX-2500/PC X射線衍射儀,日本理學公司;G9800A熒光分光光度計,美國安捷倫科技公司;NanoBrook Omni激光粒度儀,美國布魯克海文儀器公司。
1.2 兔毛角蛋白的提取
首先,將經石油醚和無水乙醇脫脂后的兔毛(RH), 采用超聲波進行預處理一定時間,振蕩頻率為50 kHz,加熱溫度為60 ℃。然后,將預處理后的兔毛纖維(UT-RH)浸泡在尿素-亞硫酸氫鈉-SDS混合溶劑體系中,調節溶劑pH值為9,加熱至90 ℃反應4.5 h。之后,將溶解液經篩孔尺寸為 0.075 mm 的篩子過濾去除未溶解兔毛得到兔毛角蛋白溶液,抽濾后采用透析管在蒸餾水中透析48 h,以去除反應中形成的小分子物質和鹽。最后,將透析后的角蛋白溶液在-80 ℃冷凍干燥獲得兔毛角蛋白粉末(RHK)。 超聲波處理時間分別為0、1、2、3、4 h, 制備的角蛋白試樣依次記為UT-0、UT-1、UT-2、 UT-3、 UT-4。
1.3 測試及表征
氨基酸含量測試:采用氨基酸分析儀對兔毛和角蛋白的氨基酸含量進行測定。將樣品置于6 mol/L 鹽酸中,于110 ℃氮氣氣氛下水解24 h。水解氨基酸由羥基琥珀酰氨基甲酸酯衍生,然后反相柱洗脫。采用高效液相色譜儀在254 nm處檢測洗脫液,用外標液(氨基酸標準溶液)通過峰面積計算樣品測定液中氨基酸的含量。
色氨酸的測定:按照GB/T 15400—2018《飼料中色氨酸的測定》,將樣品置于4 mol/L氫氧化鋰溶液中,在110 ℃氮氣氣氛下水解20 h,用乙酸鈉緩沖溶液(pH值為4.5)將水解液定量地轉移至25 mL容量瓶中,并用上述緩沖溶液定容,經過0.45 μm濾膜后利用高效液相色譜儀測定。熒光檢測器的激發波長為283 nm,檢測發射波長為343 nm,采用外標法單點校正定量色氨酸的含量。
游離巰基含量測試:根據Iesel等[12]的方法進行游離巰基的測定。將15 mg角蛋白溶解于 5 mL 的Tris-Gly緩沖液(pH值為8.0)中,然后加入50 μL Ellman試劑(DTNB溶于Tris-Gly緩沖液,質量濃度為4 mg/mL),將配好的溶液置于25 ℃條件反應60 min,離心15 min,取上清液在412 nm處測定其吸光度,以含Ellman試劑的Tris-Gly 緩沖液為空白對照。巰基含量的計算公式為
式中:A為除去空白對照后樣品在412 nm處的吸光度;D為稀釋倍數;C為樣品質量濃度,mg/mL。
角蛋白分子質量測定:利用電泳儀,采用十二烷基硫酸鈉-聚丙烯酰胺凝膠電泳(SDS-PAGE)測定兔毛角蛋白的分子質量分布[13]。RHK溶液與上樣緩沖液(0.4 g SDS,20 mg溴酚藍,2 mL甘油,1 mL 1 mol/L 的Tris-HCl(pH值為8),20 μL巰基乙醇混合定容至10 mL)以1∶1體積比混合均勻,煮沸5 min 使RHK變性。取20 μL變性RHK溶液加入預先配制好的Tris-HCl凝膠(分離膠含量為10%,濃縮膠含量為5%)中,在60 V電壓下進行電泳,當樣品進入分離膠后電壓改為120 V。電泳分離后的凝膠用考馬斯亮藍R-250染色,最后用10%乙酸和5%甲醇洗脫,直到蛋白質區帶清晰。低分子質量標準蛋白Marker在相同的電泳條件下運行。
二級結構測試:采用傅里葉紅外光譜儀(FT-IR) 對兔毛及其角蛋白的二級結構進行表征,掃描范圍為4 000~400 cm-1,掃描次數為40,分辨率為4.0 cm-1。采用Origin軟件對樣品進行基線校正、Savitsky-Golay函數平滑處理,再根據酰胺Ⅰ帶吸收峰進行歸一化處理,作其二階導數和傅里葉去卷積曲線,用高斯函數解析酰胺I帶,最后根據α-螺旋、β-折疊、β-轉角、無規卷曲各二級結構積分面積計算得出其相對含量[14]。
(5) 逐步建立適用于軌道交通行業的復合材料標準體系,包括設計標準、材料評價標準、計算驗證標準、生產工藝標準、質量檢測標準和運用維護標準等,使復合材料在軌道交通領域的應用有據可依、規范化、系統化,從而促進軌道交通行業的節能減排和綠色轉型升級。
拉曼光譜測試:采用氬離子激光器作為拉曼掃描光源進行測試,工作功率為10.2 mW,輸出波長為633 nm,光譜范圍為2 000~400 cm-1。
晶體結構測試:采用X射線衍射儀(XRD)測定兔毛及其角蛋白的晶體結構。測試條件:使用 Cu Kα 輻射源,管電壓為40 kV,管電流為200 mA,掃描方式為連續掃描,掃描速度為3 (°)/min,掃描范圍為5°~60°,步長為0.02°。利用MDI Jade軟件對樣品進行結晶度及晶面間距的計算。
三級結構測試:采用熒光分光光度計在室溫下測定角蛋白的內源性熒光光譜,激發波長為 300 nm, 掃描范圍為300~550 nm。
粒徑與電位測試:采用激光粒度儀在25 ℃條件下,對兔毛角蛋白的粒徑和電位進行測量。
2 結果與討論
2.1 氨基酸分析
兔毛及其角蛋白的氨基酸組成如圖1所示。可以看出,RH中谷氨酸(Glu)和胱氨酸(Cys)含量最多,精氨酸(Arg)、亮氨酸(Leu)、絲氨酸(Ser)和天冬氨酸(Asp)含量次之,而賴氨酸(Lys)、蛋氨酸(Met)、 組氨酸(His)和色氨酸(Trp)含量較低。與RH相比,UT-RH中大部分氨基酸組分沒有變化,但含量均有所降低。Met含量降低最為明顯,降低率高達18.50%,Cys和Lys含量降低也比較明顯,降低率分別為6.58%和5.81%;只有His、Gly和苯丙氨酸(Phe)的含量出現小幅度增加。研究顯示超聲波處理蛋白質纖維,可使纖維的細胞間質和鱗片層發生不同程度的破壞,導致水溶性蛋白溶出,造成氨基酸含量減少[15]。超聲波處理兔毛纖維的氨基酸測試結果顯示,超聲波處理不僅可使水溶性蛋白溶出,對穩定性較強的胱氨酸也有明顯的破環作用。

圖1 氨基酸含量分析結果Fig.1 Analysis result of amino acid content
與RH相比,兔毛角蛋白中胱氨酸變化最明顯,兔毛中胱氨酸含量為11.74%,提取的兔毛角蛋白中胱氨酸含量最低為2.94%,約有70%的胱氨酸被破壞。同時,超聲波處理時間不同,角蛋白胱氨酸含量也不相同,與UT-0試樣相比,UT-1、UT-2試樣的胱氨酸含量降低程度較小,當超聲波處理3 h以上,胱氨酸含量損失增大。胱氨酸作為動物纖維中重要的一種氨基酸,通過二硫鍵的結合形成高級結構,維持蛋白質纖維的形態及力學性能。提取角蛋白的關鍵是打破胱氨酸的二硫鍵,在以前的研究中顯示,超聲波處理纖維在一定程度上可使纖維的結晶度降低,這樣有利于還原劑很快進入纖維內部,與二硫鍵進行充分作用。由此可知,超聲波處理兔毛纖維有利于角蛋白的提取。此外,角蛋白中Asp、Glu、丙氨酸(ALa)、Leu、酪氨酸(Tyr)、Arg都有明顯的增加,反之Arg、Lys、脯氨酸(Pro)有所降低,且與超聲波處理時間基本呈正相關,纈氨酸(Val)、Met、異亮氨酸(Ile)、His相對較為穩定,這與氨基酸的溶解度和熱穩定性有關。最為特殊的是蘇氨酸(Thr)的變化,與RH相比,超聲波處理兔毛和角蛋白中Thr含量都有所下降,且變化基本相同,由此可知Thr幾乎不受還原體系中化學試劑的影響。
不同種類氨基酸含量結果如表1所示。可以看出,與RH相比,超聲波處理角蛋白中的總氨基酸含量降低,最高可達88.30%,這主要是由于堿性氨基酸和中性氨基酸減少,超聲波處理時間增加,這2種氨基酸損失有所降低,相反兔毛角蛋白中酸性氨基酸則有少量增加。

表1 不同種類氨基酸含量Tab.1 Contents of different kinds of amino acids
2.2 游離巰基含量分析

圖2 超聲波處理時間對兔毛角蛋白胱氨酸 及游離巰基的影響Fig.2 Effect of ultrasonic treatment on Gys and free sulfhydryl groups of keratins
提取過程中,二硫鍵在亞硫酸氫鈉的作用下斷裂形成巰基,1 mol二硫鍵斷裂生成2 mol半胱氨酸殘基,但生成的巰基不穩定,極易被氧化形成新的二硫鍵生成胱氨酸,所以游離巰基和胱氨酸的變化情況有一定差別,游離巰基隨超聲波處理時間增加大幅增加,但在超聲波處理的前2 h內,胱氨酸變化卻不明顯。巰基除再被氧化生成二硫鍵之外,有研究顯示部分游離巰基在空氣中被氧化生成砜類化合物[9],或形成揮發性硫化物,如硫化氫、揮發性有機化合物或風味物質,或者是生成硫胺酸等非常見的氨基酸,這也是游離巰基變化與胱氨酸變化不一致的原因。
2.3 分子質量及其分布分析
纖維狀角蛋白是由沿單軸平行排列的多肽鏈段組成,經過超聲波處理結合還原劑作用,二硫鍵斷裂形成分子質量不同的低硫蛋白亞基和高硫蛋白亞基。在電泳過程中,由于分子質量大小不同,這些蛋白亞基就會在電泳圖譜上分布在不同的位置[16]。采用SDS-PAGE分析了不同超聲波處理時間提取的兔毛角蛋白的分子質量,結果如圖3所示。

圖3 兔毛角蛋白的 SDS-PAGE 圖譜Fig.3 SDS-PAGE image of extracted rabbit hair keratin
電泳圖譜顯示,不同提取條件下RHK的分子質量分布不同。UT-0顯示出較為分散的條帶結構,角蛋白部分片段分子質量集中在14.4 ku以下,對應于蛋白質單體,這一系列低分子質量組分來自于嵌入中間纖維的基質中的高硫蛋白和纖維表皮;大部分片段分子質量集中在25~43 ku之間,這與纖維皮質中間絲的低硫角蛋白有關。UT-1和UT-2的分子質量分布顯示出較為分散的條帶結構,但條帶分布范圍有變窄的趨勢,大部分片段分子質量集中在31~43 ku之間,這表明從超聲波處理的兔毛中提取的角蛋白能夠解離成二聚體和三聚體。除此之外,UT-1、UT-2試樣的電泳圖譜顯示,有少量角蛋白片段分子質量集中在97.4 ku左右,且UT-2試樣的分子質量在14.4 ku以下減小。當超聲波處理時間達到3 h時,角蛋白的分子質量分布連續,且主要集中在大分子質量區域,14 ku左右的分布明顯減少。對于UT-4試樣,在整個分子質量分布區間基本都有體現,最為明顯的區間分布約在16~43 ku之間。由此可知,超聲波輔助還原法可提取大分子質量的角蛋白。
2.4 兔毛角蛋白的二級結構分析
2.4.1 化學結構分析

采用拉曼光譜儀測試了兔毛與角蛋白樣品中二硫鍵的變化,如圖4(b)所示。可知,兔毛纖維的拉曼光譜中明顯觀察到位于512 cm-1處S—S鍵的存在,超聲波處理后兔毛纖維在512 cm-1處的吸收峰強度降低,在角蛋白的光譜中S—S鍵幾乎消失,取而代之的是位于1 040 cm-1處的磺酸鹽的S—O鍵[19],這與氨基酸含量分析表現的情況一致。據資料顯示512 cm-1處振動峰來自扭-扭-扭(G—G—G)的CC—S—S—CC帶構象[20]。在不同的角蛋白樣品中,540 cm-1處出現了較為明顯的振動,該振動屬于反-扭-反(T-G-T)轉變。
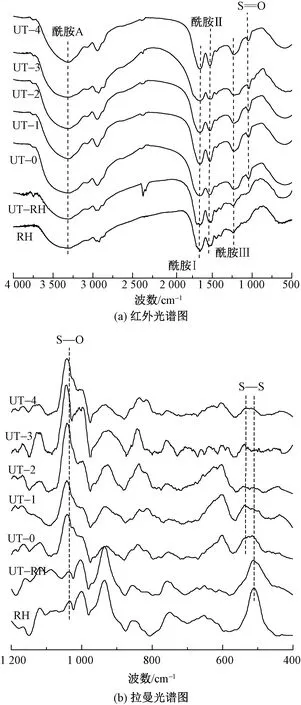
圖4 兔毛角蛋白的紅外光譜圖和拉曼光譜圖Fig.4 Infrared spectroscopy (a) and Raman spectroscopy (b) of rabbit hair keratin
紅外光譜圖中酰胺Ⅰ的吸附帶位于1 700~1 600 cm-1之間,對超分子結構特別敏感,該條帶通常用于計算α-螺旋和β-折疊結構的含量[21]。為研究超聲波作用對兔毛角蛋白二級結構的影響,對紅外光譜圖進行歸一化處理后,用高斯函數解析了酰胺Ⅰ帶,其位置、二級結構分配和相關含量百分比如表2所示。可知,兔毛纖維中α-螺旋峰面積較大(位于1 651 cm-1處),無規卷曲峰面積居中,β-折疊峰面積相對較小。隨著超聲波處理時間的增加,由于二硫鍵的斷裂,蛋白質大分子鏈被破壞,α-螺旋結構逐漸轉換為β-折疊結構和無規卷曲結構。同時,隨著超聲波處理時間增加,角蛋白的α-螺旋和β-折疊帶向較低的波數區域移動,半峰寬度值逐漸降低(即形成更窄的帶),這表明角蛋白的二級結構逐漸變得一致[22]。擬合結果還顯示,通過超聲波處理提取的兔毛角蛋白末端COOH基團含量增加,這與角蛋白提取過程中分子鏈間氫鍵、離子鍵的破壞程度增加有關[18]。
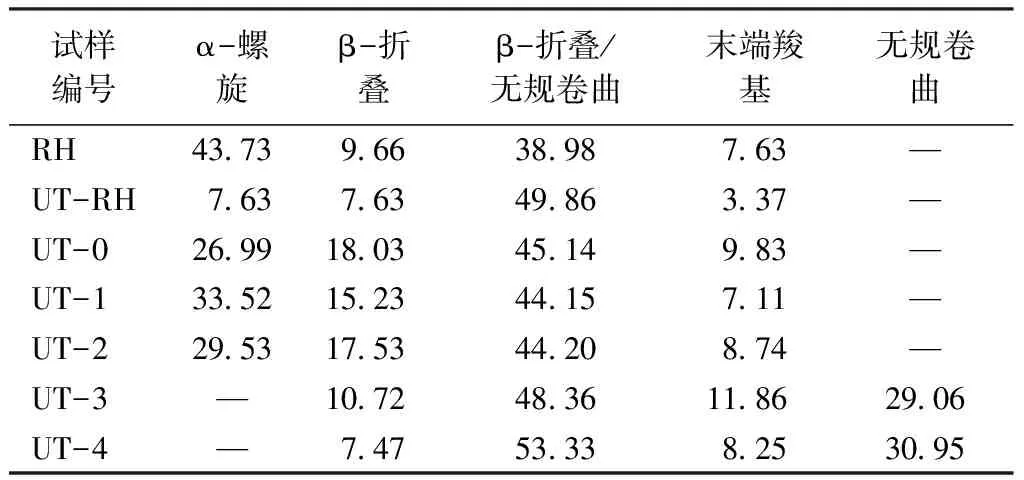
表2 兔毛及其角蛋白的二級結構含量Tab.2 Secondary structure content of rabbit hair and keratin %
2.4.2 結晶結構分析
兔毛及其角蛋白的X射線衍射譜圖如圖5所示。可知,兔毛和提取的角蛋白均在8.7°和19.12°附近出現典型的蛋白衍射峰,前者歸結為α-螺旋結晶結構衍射峰,后者歸結為β-折疊結構衍射峰[23]。兔毛纖維主要顯示出位于8.7°處的衍射峰,其結構以α-螺旋結晶結構為主。超聲波處理的兔毛纖維在8.87°處的衍射峰強度降低,而20.06°對應的衍射峰強度增加,這表明超聲波處理過程中,兔毛纖維的部分區域由于超聲波作用導致應力集中而使其結構破壞,這與紅外光譜酰胺I擬合結果一致。

圖5 兔毛及其角蛋白的X射線衍射譜圖Fig.5 X-ray diffraction for rabbit hair and keratin
與RH相比,提取的角蛋白在8.7°處的衍射峰強度明顯減弱,取而代之的是19.12°處的衍射峰強度增加,且衍射峰對應位置向低衍射角方向偏移,由20.06°偏移至19.12°左右。這主要是因為兔毛在超聲波和化學試劑的作用下二硫鍵斷裂,進而導致α-螺旋結晶結構破壞;衍射峰位置的減小表明角蛋白內2個相鄰晶格晶面間距增加,也說明超聲波處理對角蛋白晶體有膨脹作用。處理時間不同,角蛋白的結晶結構也不相同:超聲波處理2 h內,提取的角蛋白在8.7°處衍射峰的強度變化相似;當處理時間達到3 h時,該處衍射峰相對強度明顯降低,這是因為超聲波處理在一定程度上也能破壞纖維的α-螺旋結晶結構。對兔毛纖維與角蛋白的X射線衍射譜圖進行去基線后平滑,通過擬合估算了結晶度和晶格間距,結果如表3所示。可知,隨著超聲波處理時間增加,結晶度明顯降低,晶面間距逐漸增大。

表3 結晶度和晶格間距Tab.3 Crystallinity index and lattice spacing
2.5 兔毛角蛋白的三級結構分析
蛋白質的氨基酸組成中,Trp、Tyr以及Phe因具有發色基團而產生熒光。由于Phe在絕大多數實驗條件下不被激發,Tyr殘基又會將吸收的能量轉移給Trp殘基,使得Trp殘基的熒光增強,所以角蛋白的內源熒光主要來自Trp[24]。Trp屬于疏水性氨基酸,微環境的變化對其熒光光譜影響很大,通過色氨酸殘基發出的內源熒光光譜可反映出蛋白質結構的變化。兔毛角蛋白的內源熒光光譜如圖6所示。可知,提取的兔毛角蛋白具有熒光特征。角蛋白的氨基酸含量測試結果顯示色氨酸占0.68%,酪氨酸占4.61%,苯丙氨酸約占3.53%,這3種氨基酸的分子結構存在能夠發色的熒光基團——吲哚環和苯基殘基。不同超聲波處理條件下提取的角蛋白的熒光特征峰位置也不相同。其中,UT-0、UT-1位于 358 nm 處,UT-2和UT-4位于356 nm處,UT-3位于354 nm處,但均處在(356±2) nm范圍內,這與Trp的熒光發射光譜相同。隨著超聲波處理時間的增加,提取的角蛋白熒光發射譜發生很小的藍移,熒光強度小幅增加。蛋白質熒光光譜受溶劑極性的影響較大,極性環境會影響生色基團的基態和激發態能級,減少激發態的能量,從而引起發射譜的紅移,強度降低[25]。由此可知,超聲波處理時間越長,提取的角蛋白中芳香族氨基酸分子所處的局部小環境的極性越小,弱于角蛋白分子外部水溶液的極性,其被深埋于角蛋白的內部,被多種非極性氨基酸殘基包圍。
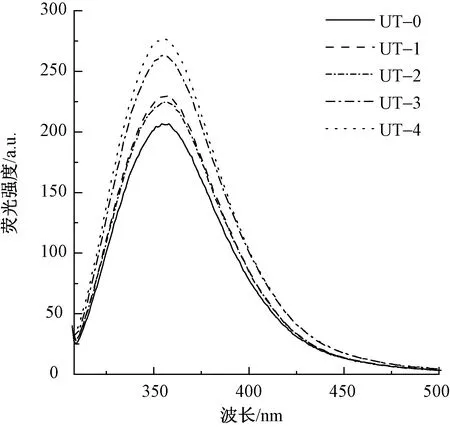
圖6 兔毛角蛋白的內源熒光光譜Fig.6 Endogenous fluorescence spectra of rabbit hair keratin
2.6 兔毛角蛋白粒徑和電位分析
蛋白質溶液的穩定性和聚集程度可通過平均粒徑和ζ-電位反應出來,超聲波處理時間對角蛋白粒徑分布的影響如圖7(a)所示。可知,隨著超聲波處理時間的增加,角蛋白的粒徑表現出不同程度的偏移,超聲波處理時間在2 h以內時,角蛋白粒徑分布明顯右移,而當超聲處理時間增加到3 h后,粒徑分布又出現了一定的左移。UT-0、UT-1、UT-2、UT-3和UT-4的平均粒徑分別為164.22、179.64、178.81、159.19 和158.53 nm。

圖7 兔毛角蛋白的粒徑分布與ζ-電位值Fig.7 Particle size(a) and ζ-potential(b) of rabbit hair keratin
不同超聲波處理時間下提取的兔毛角蛋白的 ζ-電位如圖7(b)所示。可知,超聲波處理會使提取的角蛋白的ζ-電位絕對值降低,當處理時間在3 h以內時ζ-電位值基本相近,超聲波處理時間增加到4 h時,ζ-電位絕對值降低明顯。這說明隨著超聲波處理時間增加,提取的角蛋白表面凈電荷數減少,體系開始變得不穩定。
角蛋白的粒徑大小受到大分子鏈斷裂程度、分子間聚集行為以及大分子鏈伸展情況的綜合影響。研究顯示超聲波處理時間越長,纖維溶解率越大,此時大分子鏈之間破壞程度增加[26],這可能會導致提取的角蛋白的粒徑減小。同時由熒光測試可知,超聲波處理時間越長,疏水性的芳香族氨基酸處于蛋白質內部,角蛋白分子鏈伸展程度越低,所以粒徑越小。但角蛋白具有良好的自組裝特性,ζ-電位測試結果顯示,處理時間增加時提取的角蛋白不穩定,此時角蛋白分子間依靠氫鍵、離子鍵等相互集聚,又會使粒徑增大,所以角蛋白粒徑變化與超聲波處理時間變化并不完全相同。
3 結 論
本文詳細研究了兔毛角蛋白的化學組成及各級結構,研究結果表明采用超聲波輔助還原法提取的角蛋白中氨基酸含量可達88.30%。隨著超聲波處理時間增加,胱氨酸損失增大,游離巰基呈先增加后減小的趨勢,高分子質量蛋白的相對含量增加,分子質量集中分布在31~43 ku之間和97.4 ku以上,有利于角蛋白的再生利用。角蛋白顯示出典型的蛋白質結構,隨著超聲波處理時間的增加,α-螺旋結構逐漸減少,轉換為β-折疊和無規卷曲結構,且峰值向較低的波數區域移動,結晶度顯著降低。提取的角蛋白具有穩定的三級結構,角蛋白分子不易發生集聚,形成穩定的角蛋白溶液。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
