過渡金屬催化的6-苯基-5,6-二氫-2H-吡喃-2-酮的合成
——推薦一個大學有機化學綜合實驗
2022-03-30 01:31:48劉治彤趙子逸彭渤曲玥名田松麟俞壽云
大學化學 2022年2期
劉治彤,趙子逸,彭渤,曲玥名,田松麟,俞壽云
南京大學化學化工學院,南京 210023
1 引言
碳-碳鍵的形成和斷裂是有機合成的中心問題。科學家們通過不斷的探索研究,已經做出了許多偉大的成就。本次實驗以碳-碳鍵連接為中心議題展開,通過兩種不同的碳-碳鍵連接方式進行三步合成反應。可以在實驗過程中體會碳-碳鍵形成在有機合成中的重要地位,并且了解經典的和相對前沿的碳-碳鍵形成的多種方式。
金屬介導或者催化的反應是形成碳-碳鍵的強有力方法,該類反應備受矚目。在這個領域曾有過三次諾貝爾獎級的成果,分別是1912年的“格氏反應”、2005年的“烯烴復分解反應”以及2010年的“鈀催化的交叉偶聯反應”。格氏反應已經進入本科生理論和實驗的學習,但在實驗過程中,存在格氏試劑較難引發、引發現象不很明顯等一系列困難,因而,本實驗提出一種改進的方式,更好地完善格氏反應的實驗教學。目前已有部分國內外高校在本科化學實驗中開設了鈀催化的交叉偶聯反應的實驗教學內容[1]。迄今為止,烯烴復分解反應在本科理論教學中少有涉及,實驗教學中更是缺少這一諾獎級的成果。因而,本實驗設計了一種適用于本科教學的烯烴復分解反應,有利于學生更好地全面了解有機化學的重要發展歷程和成果。
最為經典的碳-碳鍵的形成反應當屬格氏反應。1912年,諾貝爾化學獎授予了Victor Grignard。Grignard借鑒了Frankland和Wanklyn制備有機鋅試劑的方法,在室溫、常壓下成功完成了鎂試劑的制備并和酮反應制備醇,該成果于1900年發表[2]。格氏反應已經較為成熟,在基礎有機化學實驗中多有涉及。但是基礎實驗教學中經典的格氏試劑引發過程不是很容易,且不易判斷。本實驗嘗試改進,通過反應溶液顏色的明顯變化指示反應的進程。
烯烴復分解反應是前沿的碳-碳鍵形成反應,是連接sp2-碳和sp2-碳的高效方法。從反應的凈結果來看,是兩個烯烴在催化劑的作用下相互交換,因而也被形象地譽為“交換舞伴的反應”。烯烴復分解反應是2005年諾貝爾化學獎的成果。最初,該反應在工業界被發現[3]。如今,烯烴復分解反應,特別是關環復分解反應(Ring Closing Metathesis,RCM),為眾多天然產物的合成提供了便捷的路徑[4]。學習并深入了解該反應,可以拓寬有機合成的思路。最初烯烴復分解反應被發現時,雖然在有機合成以及工業界有巨大的應用潛力,但是催化劑體系對于水和氧氣不穩定,限制了其推廣。Richard R.Schrock教授和Robert H. Grubbs教授研究并且發明了更加高效穩定的催化劑,為烯烴復分解反應的應用做出了巨大貢獻。
Schrock教授于1980年發現了鉭卡賓配合物具有烯烴復分解的能力[5]。隨后Schrock教授所研發的鉬或鎢的催化劑有更好的反應活性[6],其中圖1A中的催化劑已經商品化。Schrock教授的工作使得烯烴復分解反應的推廣成為可能,為后續新一代催化劑的出現打下了堅實的基礎。雖然鉬或鎢的催化劑活性較好,但是,對于空氣和水仍然很不穩定。1992年,Grubbs教授發現釕卡賓金屬化合物能夠進行降冰片的開環聚合反應,且該催化劑能夠對水、空氣等穩定[7]。釕催化劑的出現為烯烴復分解反應打通了另外一條道路。該催化劑見圖1B。1996年,Grubbs對釕催化劑進行改進,成為了第一代Grubbs催化劑,其催化性能相較于之前的催化劑更好,其結構式見圖1C[8]。隨著研究的深入,1999年,Grubbs發現催化劑的活性與其膦配體的解離有關,認為催化循環過程中經過一個高活性的單膦中間體,從而根據這樣的事實發明了第二代催化劑,其結構式見圖1D[9]。催化劑用量更少,適用于開環復分解以及關環復分解反應。

圖1 常用的烯烴復分解催化劑
本實驗以6-苯基-5,6-二氫-2H-吡喃-2-酮(6)作為目標化合物。使用Cp2TiCl2催化的烯丙基溴(1)對苯甲醛(2)的加成反應,4-二甲氨基吡啶(DMAP)催化的醇(3)與丙烯酰氯(4)的酯化反應和Grubbs II催化劑催化的烯烴(5)的關環復分解反應作為關鍵反應,合成吡喃酮(6)。實驗步驟如圖2所示。

圖2 6-苯基-5,6-二氫-2H-吡喃-2-酮的合成的反應步驟
2 實驗目的
(1)通過查閱文獻,了解過渡金屬催化有機反應的歷史和現狀。
(2)熟悉并了解Cp2TiCl2催化的醛的加成反應,DMAP催化的酯化反應和烯烴復分解反應的原理。
(3)掌握簡單無水無氧的操作。
(4)掌握利用TLC監測有機反應和利用快速柱層析純化反應產品。
(5)學會通過核磁共振譜圖鑒定反應產物。
(6)鞏固減壓過濾、萃取、分液等基本有機實驗操作。
(7)掌握旋蒸儀的使用方法。
3 試劑和儀器
3.1 反應試劑
二氯二茂鈦,4-二甲氨基吡啶(DMAP),Grubbs II催化劑(其結構式見圖1D)。以上試劑采購自畢得醫藥。烯丙酰氯,無水四氫呋喃(有分子篩),二氯甲烷(分析純),乙酸乙酯(EA,分析純),石油醚(PE,分析純)。以上試劑采購自安耐吉。其余試劑還包括:烯丙基溴(國藥試劑),苯甲醛(阿達瑪斯試劑),活化鋅粉(自制),三乙胺(上海泰坦化學有限公司),高純氮氣(南京寧衛醫用氧氣有限公司),無水二氯甲烷(有分子篩,百靈威),飽和氯化銨溶液(其中,氯化銨采購自西隴化工股份有限公司),飽和食鹽水(其中,氯化鈉采購自西隴科學股份有限公司),乙醚(分析純,南京化學試劑股份有限公司),無水硫酸鎂(永華科學科技有限公司),磷鉬酸溶液(配制方法:10 g磷鉬酸+ 100 mL乙醇)。
3.2 實驗儀器
分析天平,磁力攪拌器,紫外燈(254 nm),旋轉蒸發儀,400 MHz核磁共振儀(CDCl3為氘代試劑,TMS為內標物)。儀器的型號、廠家和國家見表1。

表1 實驗儀器匯總
4 實驗步驟
4.1 Cp2TiCl2催化的烯丙基溴對苯甲醛的加成反應[10]
準備一個100 mL梨形瓶,在其磨口上用翻口橡膠塞塞緊并用密封膠帶在橡膠塞與瓶口的接觸部位纏繞數圈。用雙排管抽換氣3次并充入氮氣。準備好兩個氮氣球。將上述準備好的氣球插在置換好氣體的梨形瓶橡膠塞上,用1 mL注射器在上述梨形瓶中加入苯甲醛(6 mmol,0.64 g),烯丙基溴(15 mmol,1.81 g)并用注射器加入30 mL無水四氫呋喃,配制成待用溶液。
在另一個100 mL梨形瓶中放入適當大小磁子,加入二氯二茂鈦(0.06 mmol,14.9 mg)和活化鋅粉(15 mmol,0.98 g)。磨口處塞上翻口橡膠塞并用密封膠帶纏繞數圈,通過針頭和雙排管進行抽氣以及氮氣置換,此步驟重復三次后,用氮氣保護體系,并插上氮氣球。室溫下開啟磁力攪拌,并用注射器加入無水四氫呋喃30 mL(反應裝置見圖S1)。溶液顏色成為紅色,等待體系顏色變綠后(大概需要10 min左右) (第一步反應顏色變化見圖S2),注射入之前配制好的苯甲醛和烯丙基溴混合溶液。反應持續20 min。用TLC監測反應是否完成(原料點使用苯甲醛;展開劑可以用V(PE) :V(EA) = 9 : 1)。若未反應完成,繼續反應10 min,再次使用TLC監測反應是否完成。在實驗報告本上按原比例大小繪制TLC分析結果并分別計算Rf值(該反應的TLC分析結果見圖S3)。反應的化學方程式如圖3所示。
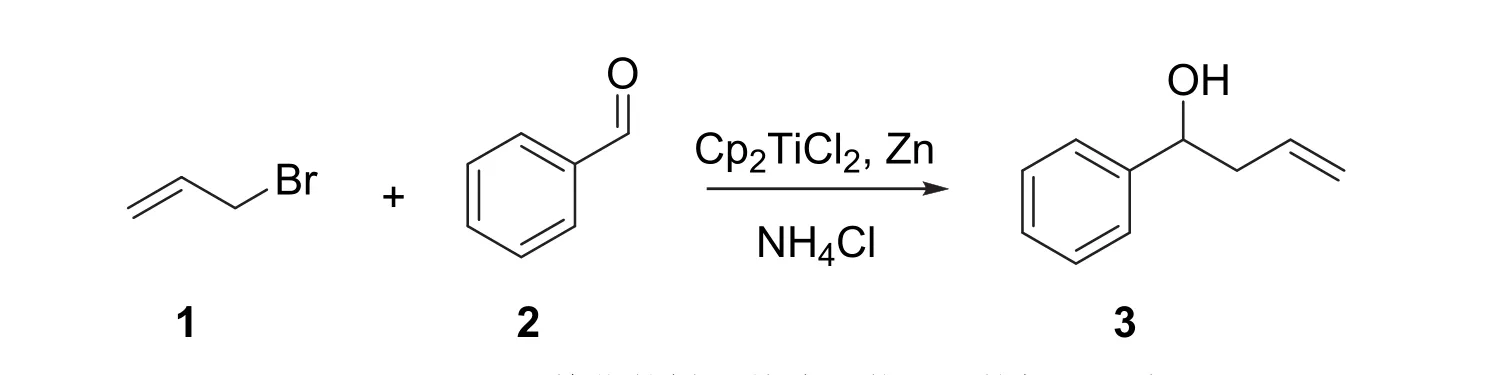
圖3 Cp2TiCl2催化的烯丙基溴對苯甲醛的加成反應
反應結束后,拔去橡膠塞,緩慢滴加飽和氯化銨溶液20 mL淬滅反應。減壓過濾除去沉淀,得到的溶液用20 mL乙醚分層,在分液漏斗中分離水相和有機相,水相再用乙醚萃取三次,每次用10 mL,合并有機相。有機相再用飽和食鹽水洗三次,每次用10 mL。洗過后的有機相用無水硫酸鎂干燥。過濾去除固體沉淀,溶液接在一個250 mL的茄形瓶中。調整旋轉蒸發儀的水浴溫度為35 °C左右,將茄形瓶裝在旋轉蒸發儀上并用塑料夾固定。開啟泵后調節壓力并旋轉旋鈕使體系密封,先緩慢開啟旋轉,待溶劑開始蒸出且沒有爆沸后,將茄形瓶放入水浴中加速旋轉,快速蒸除溶劑。
使用快速柱層析純化產物。淋洗液使用V(PE) :V(EA)從50 : 1到20 : 1到10 : 1梯度洗脫。產物為無色油狀液體3。稱重,計算產率(0.78 g,產率88%)。用NMR鑒定,譜圖見補充材料圖S4和S5。1H NMR (400 MHz, CDCl3):δ7.35-7.25 (m, 5H),5.86-5.75 (m, 1H),5.19-5.11 (m, 2H),4.74-4.71(dd,J= 4.8, 8.0 Hz, 1H),2.56-2.45 (m, 2H)。13C NMR (101 MHz, CDCl3):δ143.93,134.51,128.40(2C),127.56,125.86 (2C),118.38,73.35,43.83。
4.2 DMAP催化的醇與酰氯的酯化反應(圖4) [11]
在25 mL兩頸瓶中加入適當大小磁子,加入DMAP (0.2 mmol,24.4 mg),在兩頸瓶上磨口處連接滴液漏斗,將滴液漏斗上方和兩頸瓶另一瓶口用翻口橡膠塞塞住并用密封膠帶纏繞數圈,進行氮氣置換并用氮氣保護體系。橡膠塞上插入氮氣球后,室溫攪拌下依次從兩頸瓶側瓶口處注射加入5 mL無水二氯甲烷,第一步產物3 (4 mmol,0.59 g),三乙胺(8.8 mmol,0.89 g)。在滴液漏斗中注射酰氯4 (8 mmol,0.72 g),冰浴下小心緩慢滴加酰氯入反應體系,在20-30 min內滴加完成(反應裝置見圖S6)。撤去冰浴,室溫下反應3 h,用TLC分析反應是否完成(該反應的TLC分析結果見圖S7)。(展開劑可以用V(PE) :V(EA) = 9 : 1)如果沒有完成可以繼續反應20 min后監測。反應的化學方程式如圖4所示。

圖4 DMAP催化的醇與酰氯的酯化反應
反應結束后,拆除滴液漏斗等裝置,緩慢滴加10 mL飽和食鹽水淬滅反應,轉移至分液漏斗中并用5 mL二氯甲烷分層,水相用二氯甲烷萃取三次,每次5 mL。合并有機相,并用飽和食鹽水洗滌三次,每次5 mL。有機相用無水硫酸鎂干燥,過濾固體后,溶液用旋轉蒸發儀除去溶劑。使用快速柱層析純化產物。淋洗液使用V(PE) :V(EA)從50 : 1到20 : 1到10 : 1梯度洗脫。產物為無色液體5。稱量,計算產率(0.58 g,產率69%)。用NMR鑒定,譜圖見補充材料圖S8、S9。1H NMR (400 MHz, CDCl3):δ7.35-7.26 (m, 5H),6.45-6.40 (dd,J= 1.6, 16.8 Hz, 1H),6.19-6.11 (dd,J= 10.0, 17.2 Hz, 1H),5.90-5.86 (dd,J= 5.6, 7.6 Hz, 1H),5.84-5.81 (dd,J= 1.6, 10.4 Hz, 1H),5.77-5.66 (m, 1H),5.11-5.03(m,2H),2.74-2.56 (m, 2H)。13C NMR (101 MHz, CDCl3):δ165.33,139.98,133.21,130.85,128.60 (2C),128.44,127.98,126.53 (2C),118.11,75.34,40.76。
4.3 Grubbs II催化劑催化的烯烴關環復分解反應(圖5) [12]
在150 mL兩頸瓶中加入適當大小磁子和Grubbs II (0.056 mmol,47.5 mg,結構式見圖1D),兩頸瓶上磨口處連接上球形冷凝回流管后,所有接口處塞上翻口橡膠塞并用密封膠帶密封。體系置換氮氣并用氮氣球保護。在兩頸瓶側瓶口處加入無水二氯甲烷60 mL,加入第二步反應產物(0.56 mmol,0.11 g) (反應裝置見圖S10)。在45 °C下攪拌4 h。用TLC分析(展開劑可用V(PE) :V(EA) = 5 : 1) (該反應的TLC分析結果見圖S11)。反應的化學方程式如圖5所示。
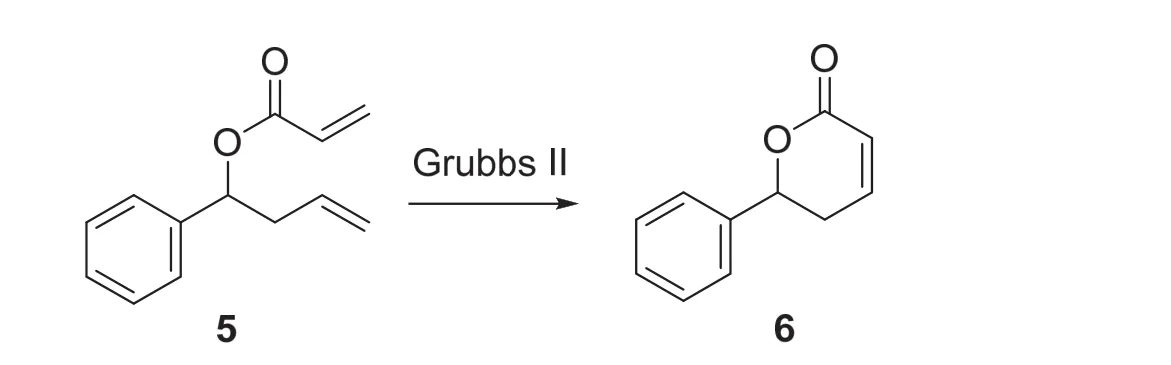
圖5 Grubbs II催化劑催化的烯烴關環復分解反應
用10 mL飽和食鹽水淬滅反應,10 mL二氯甲烷分層,水相用二氯甲烷萃取三次,每次5 mL,有機相用飽和食鹽水洗滌三次,每次5 mL。有機相用無水硫酸鎂干燥,過濾固體后旋蒸溶劑,使用快速柱層析純化產物。淋洗液用V(PE) :V(EA)從5 : 1到3 : 1梯度洗脫。產物為白色固體6 (0.07 g,產率73%)。稱重,計算產率。并用NMR鑒定,譜圖見補充材料圖S12、S13。1H NMR (400 MHz, CDCl3):δ7.43-7.33 (m, 5H),6.99-6.94 (ddd,J= 3.2, 5.6, 9.6 Hz, 1H),6.16-6.12 (ddd,J= 1.2, 2.0, 9.2 Hz, 1H),5.48-5.43 (dd,J= 5.2, 11.2 Hz, 1H),2.72-2.57 (m, 2H).13C NMR (101 MHz, CDCl3):δ164.01,144.82,138.47,128.69 (2C),128.64,126.05 (2C),121.77,79.25,31.60。
4.4 實驗注意事項
(1)實驗中涉及用雙排管的抽換氣過程,雙排管裝置圖、制作氮氣球、移取無水溶劑的操作具體細節見補充材料圖S14-S16。
(2)本綜合實驗反應均在無水無氧的體系下進行,因此在用注射器加入液體的過程中,盡量減少將空氣注射入反應體系,導致對實驗結果造成較大的影響。
(3)薄層層析技術以及柱層析技術具體細節見補充材料中操作指南細節解說。
(4)酰氯對水和空氣很不穩定,須在通風櫥內操作,盡量減少其暴露在空氣中的時間,并且需保證反應儀器充分干燥,不能有水殘留,在稱量酰氯時可以先在通風櫥內加在小號塑料離心管中,封好塑料離心管蓋子后去稱量臺稱量。
(5)第二步反應體系溶液分層不易,在萃取洗滌過程中會出現乳化現象,等待時間較長,必要時可以進行離心操作;該步反應有機相在下層。
(6)柱層析中梯度洗脫的方法為:先用小極性的淋洗液沖洗過柱至雜質點先被洗脫出來,之后換用極性更大的淋洗液進行淋洗,使得所需要的產物快速從硅膠柱中洗脫下來。
5 結果和討論
5.1 Cp2TiCl2催化的烯丙基溴對苯甲醛的加成反應
格氏反應是高效形成碳-碳鍵的方法之一,但是一般傳統的格氏反應引發較為困難,對于不同的底物,引發所需要的溫度條件等不盡相同,且是否引發成功較難判斷,反應的總時間也相對較長。在反應引發階段常會加入碘與鹵代烴的鹵素原子進行交換,便于引發。2012年,Brandon L.Ashfeld教授嘗試選擇一種催化劑,與鹵代烴發生氧化加成,從而制備出金屬有機化合物,進行后續碳-碳鍵的形成[10]。具體機理見圖6。在催化循環的過程中,首先是金屬(本實驗中為鋅)將二氯二茂鈦中的四價鈦還原至三價,從而三價鈦可以還原鹵代烴,斷裂C―X鍵,生成C―Ti鍵。接著,Zn再次將四價鈦還原至三價,以便可以和二價鋅發生轉金屬過程得到有機鋅試劑,最后完成與羰基化合物的親核進攻得到終產物。而三價鈦則進入下一次循環。

圖6 二氯二茂鈦催化循環的可能機理
該方法的實驗現象非常明顯,加入二氯二茂鈦,使得反應體系的顏色成紅色,當引發完成后,體系成為綠色,通過目測即可判斷引發是否完成。總共只需要半小時就可以完成90%左右的轉化。催化劑二氯二茂鈦的用量僅為1%-2% (摩爾分數),且較易儲存,不易變質,價格也不是很昂貴,適合本科實驗教學。
重復實驗的過程中發現該反應引發的時間與反應物的量有一定的關系,隨著反應物增多,引發所需要的時間相應延長。但由于實驗現象明顯,即發生顏色的改變,可以根據這一實驗現象進行判斷,適當增多或減少原料的用量。
溫度和季節的變化對于該反應也有一定的影響。在空氣濕度大的季節重復實驗,所得到的結果產率會稍微偏低,在做實驗的過程中就要注意抽換氣的次數可以更多一些,盡量讓反應體系中少一些空氣以及水蒸氣。隨著溫度的升高,引發過程逐漸變快,在剛加入溶劑,即無水四氫呋喃的時候就可以看見溶液呈現較淺的綠色。為了使反應完全,引發過程至少不少于5 min。
該步反應進行多次重復,收率范圍在84%-92%之間。
5.2 DMAP催化的醇與酰氯的酯化反應[11]
最常見的酯化反應通常是由羧酸和醇在強酸催化下進行反應,但該反應為可逆反應,難以轉化完全。需要通過其他途徑,如不斷移除產物或加入過量酸、醇等方法提高產率。除此以外,還可以通過酰氯或酸酐與醇進行反應,由于酰氯和酸酐的反應活性大于羧酸,用酰氯或酸酐反應,可以更好地提高轉化率,得到更多預期產物。本實驗采用酰氯和醇進行第二步酯化反應,以期更快完成,產率也更高。
該反應使用DMAP作為催化劑。DMAP上有兩個氮原子,其中三級胺上的氮原子可以將自己的孤對電子共振到吡啶環上,而吡啶環上氮原子的孤對電子相對定域,使得吡啶環上氮原子的親核性增強。DMAP吡啶環上的氮原子比醇羥基上氧原子的親核性更強,DMAP作為催化劑的催化機理如圖7所示:DMAP作為親核試劑進攻酰氯而生成酰基吡啶陽離子,醇進攻酰基吡啶陽離子的羰基得到產物酯,并同時得到質子化的DMAP。經過NEt3堿處理,再生的DMAP進入下一次的催化循環[13]。
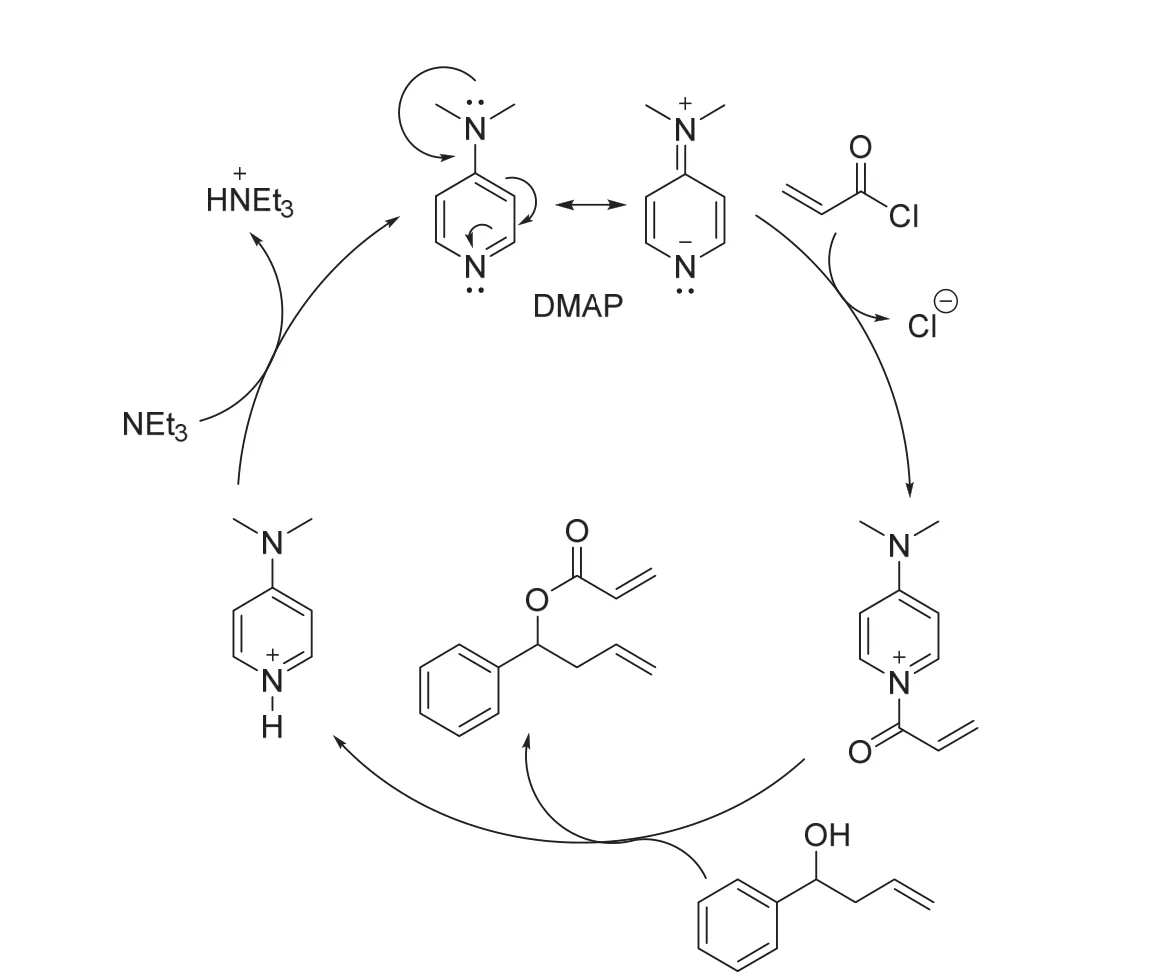
圖7 DMAP催化酯化反應機理
在該步反應過程中,由于體系成堿性,生成的酯可能在該環境下不穩定,因而會對最終的收率有一定的影響。如果反應完成后長時間不處理,可能造成一部分產物的水解,又生成醇,使產率下降,同時也對柱層析分離造成一定麻煩,因而該反應完成后需要迅速淬滅。該步反應使用高活性的酰氯,如果實驗過程中對于恒壓滴液漏斗的控制不當造成酰氯過快加入到反應體系中,會導致放熱過快而不利于反應。根據這一特點,加入酰氯時也可以選擇用注射器進行人工控制滴加酰氯,使反應體系更加穩定。
該步反應對水十分敏感,應盡量保證體系干燥,可以通過多次抽換氣達到這一目的。原料醇也需要保持干燥無水,如果在空氣中存放時間過久,可以用少量乙酸乙酯溶解重新旋蒸并抽干再進行反應。如果實驗室提供的三乙胺為分析純的試劑,可以直接使用,也可以采購無水的三乙胺來完成實驗。為減小水對于反應的影響,可以多加入一點酰氯,但結果會稍有偏差。
該步反應進行多次重復,收率范圍在64%-70%之間。
5.3 Grubbs II催化的烯烴關環復分解反應[12]
Yves Chauvin教授和他的學生在1971年提出了烯烴復分解的反應機理,該機理被許多實驗結果所證實,如今廣被有機化學家所接受[14]。Chauvin教授也因此成為2005年諾貝爾化學獎得主之一。具體反應機理見圖8。反應首先是由催化劑中的金屬(本實驗中為釕)-碳雙鍵和原料中的其中一根碳碳雙鍵進行[2 + 2]環加成,生成四元環中間過渡態。接著經歷一個開環過程,得到釕卡賓物種。該中間體再次和原料中另一根碳碳雙鍵發生[2 + 2]環加成,經歷四元環過渡態后,開環得到目標產物。在本實驗中使用的是Grubbs第二代催化劑。
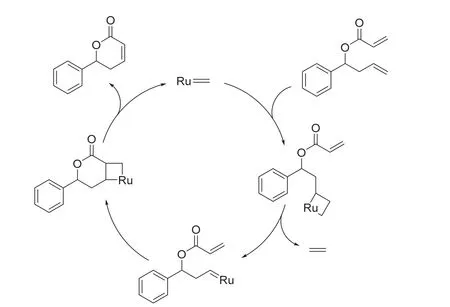
圖8 烯烴復分解反應機理
在第三步反應的摸索過程中,發現反應溶劑和濃度對該反應的影響很大,具體結果如表2所示。使用二氯甲烷作為溶劑,反應完成需要4 h左右。作為教學實驗,不特意強調產率的情況下,可以縮短反應時間。TLC分析結果干凈,只有產物點。終產物在二氯甲烷以及乙酸乙酯中溶解度較好,旋蒸溶劑時不易出現固體,可用石油醚重結晶來獲得產物進行后續分析實驗。

表2 烯烴關環復分解反應的條件優化
該步反應進行多次重復,收率范圍在73%-82%之間。
6 教學組織運行方式
本實驗在理論上涉及新的知識較多,需要學生在實驗前充分查資料了解實驗的一些重要反應,如DMAP為催化劑的酯化反應、烯烴復分解反應等。只有在實驗前充分理解這些原理,在實驗過程中才會有更多收獲。同時,也可以專門在實驗正式開始前設置一節理論課,梳理一下實驗中的重要知識點,開拓一下學習有機化學的思維。
實驗中涉及的新操作有通過雙排管進行無水無氧處理、用注射器移取液體樣品、無水液體樣品的注射、旋轉蒸發儀的使用等等,這些實驗操作在原來本科教學實驗中較少出現,但在科研過程中非常有用,學習這些有用的技能是很必要的,因此也需要花一定的精力完成這類操作的教學,可以通過錄制視頻讓學生提前觀看后學習或者在實驗過程中教師示范等方式進行。
本綜合實驗中后兩步的反應時間過長,在實際的教學中可以由教師先錄制好實驗操作重點的視頻,學生提前觀看預習,并且在實驗當天由教師提前稱量好藥品,學生到實驗室直接搭建反應裝置開始反應,在反應的過程中完成實驗原理的講解。也可以適當縮短反應時間,但反應收率會相應降低。
最后一步RCM反應在本次綜合實驗中操作難度較大,容錯率較低,稍有疏忽就會使得產率下降。在實驗中需要著重關注水對于反應體系的影響,無水條件越好,產率會相應越高。可以將最后一步反應設定為本次綜合實驗的考核反應,檢驗學生在“模擬”科研實驗室條件下的實驗水平。該步反應的產物為固體,在最終柱層析的時候可以考慮使用干法裝柱、干法上樣,會縮短一定的實驗時間,同時也多方面訓練了學生的實驗技巧。
完成第一步實驗需要約4-6 h,完成第二步實驗需要約7-8 h,完成第三步實驗需要約7-8 h。該合成的每一個反應都可獨立成為一個基礎有機化學實驗,三步的連續反應可以作為一個綜合多步合成實驗。
7 創新性/特點/特色聲明
(1)可視化金屬有機試劑的制備;
(2)烯烴復分解反應的引入;
(3)有機化學相關的模擬科研實驗訓練。
8 結語
本實驗以有機合成中重要的碳-碳鍵形成為主要研究對象進行展開。第一步反應改進了格氏反應引發困難以及不易判斷引發成功與否的問題,使用Cp2TiCl2作為催化劑,更快更容易地形成有機鋅試劑,并可目測判斷,以較高的收率得到產物。其次,通過酯化反應,為第三步烯烴復分解反應做好準備。烯烴復分解反應作為諾貝爾化學獎級的成果,如果可以進入本科理論教學乃至實驗教學的課程中,有利于學生對于最新化學進展的認知與了解。整個綜合實驗中不僅包含有機合成的常規訓練,還增添了許多貼近科研的實驗操作。可以訓練學生的實驗技能和動手能力,有助于學生對有機化學的認識和理解。
補充材料:可通過鏈接http://www.dxhx.pku.edu.cn免費下載。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
