核磁共振技術測定聯萘酚的對映體純度
2022-03-30 01:31:48奚忠華陸大東戴潔葉濤章文偉
大學化學 2022年2期
奚忠華,陸大東,戴潔,葉濤,章文偉
化學國家級實驗教學示范中心(南京大學),南京 210023
在不對稱合成和新藥的研發過程中,測量手性化合物的純度(對映體純度)是一項必須的工作。通常使用對映體過量(enantiomeric excess,簡稱ee)來衡量手性化合物的純度。實驗室中測定ee值常用的方法是手性固定相高效液相色譜法(chiral HPLC)。采用手性色譜方法來測定ee值往往需要消耗大量時間,難于滿足高通量的測試需求。核磁共振技術是鑒定有機化合物結構的有效手段,采集一個核磁氫譜消耗的時間小于五分鐘。相較耗時的手性色譜方法,核磁共振法可以更加快速地測定化合物的ee值。
手性化合物中對映的氫核所產生的磁共振信號頻率相同,這意味著其在核磁共振譜圖中的化學位移相同,因此無法通過核磁共振信號來區分對映體。通過添加手性試劑,可以將對映體化合物轉變成非對映體的混合物,混合物中原對映氫核若能產生適合分辨的信號,則可以根據信號的強度來確定原對映體化合物中對映體的含量,從而得到ee值[1]。
在醫藥工業中,高純度手性化合物有著廣泛的應用,因此手性合成是有機化學研究的熱點領域之一。在各種手性合成方法中,不對稱催化是一種非常高效的方法。以手性聯萘酚((±)-binaphthol,(±)-BINOL) (2) (圖1)及其衍生物為配體的金屬配合物是應用廣泛的一類手性催化劑,因而在教學中開設手性聯萘酚的制備實驗[2]是引入手性合成的有效手段,目前全國很多高校都開設此實驗。手性聯萘酚制備完成后必須進行光學純度的測定,以檢驗合成方法的有效性。常見的測定方法有液相色譜法[3,4]、毛細管電泳法[5]和核磁共振法[6,7],本文對于手性聯萘酚的ee值測定采用簡便的三組分手性衍生方法[8]。聯萘酚與2-甲酰基苯硼酸(2-FPBA) (1)和(S)-(-)-1-苯乙胺((S)-MBA) (3)三者混合時會發生Bull-James Assembly反應[9],得到亞氨基硼酸酯(4)的非對映體混合物,其中(R,S)-4和(S,S)-4的含量比就對應了(±)-BINOL中不同構型的比例(圖1)。這種手性衍生試劑核磁共振方法還可用于手性1,2-,1,3-,1,4-二醇和十多種手性伯胺的ee值測定[10-12]。

圖1 測定聯萘酚ee值的Bull-James Assembly反應過程
1 實驗
1.1 儀器與試劑
核磁共振波譜儀(布魯克AVANCE III 400M,瑞士),高效液相色譜儀(島津LC20-AT,日本,二元泵,紫外檢測器),純水機(AWL-6000-U,艾科浦),移液器。
2-FPBA (99.72%,上海皓鴻生物醫藥科技有限公司),(S)-MBA (99%,薩恩化學技術有限公司),氘代氯仿(99.8%,0.03%TMS,薩恩化學技術有限公司),(R)-BINOL (99%,上海笛柏生物科技有限公司),(S)-BINOL (99%,上海笛柏生物科技有限公司),聯萘酚學生產品,分子篩(4A型,國藥集團化學試劑有限公司),甲醇(HPLC級,TEDIA),超純水。
1.2 樣品準備
分別準確稱取29.0 mg BINOL標準品((R)-BINOL含量范圍為90%-10%)和15.0 mg 2-FPBA,轉移至樣品管中,用移液器加入2.00 mL氘代氯仿溶解樣品。將樣品管放入溫水浴中,加快溶解速度。溶解完成后,加入4A分子篩5-6顆覆蓋樣品管底,干燥,待用,記為溶液A。學生產品采用同樣操作步驟。
準確稱取37.0 mg (S)-MBA于10.0 mL樣品管中,用移液器加入5.00 mL氘代氯仿,混合均勻后加入4A分子篩5-6顆覆蓋樣品管底,干燥,待用,記為溶液B。
1.3 液相色譜分析
用移液器移取干燥后的溶液A 100 μL于2 mL樣品管中,再加入900 μL甲醇稀釋,取5 μL進樣分析。液相色譜分析條件如下:采用菲羅門Chirex (S)-VAL and DNAn手性分離柱(4.6 × 250 mm,5 μm),柱溫40 °C,流動相甲醇-水(體積比70 : 30),流速1.0 mL·min-1,檢測波長262 nm。
1.4 核磁分析
用移液器分別移取干燥后的溶液A和溶液B各300 μL于同一核磁管內,混合均勻后進行1H NMR測試。核磁譜圖采集使用脈沖序列為zg,掃描次數為16次,探頭溫度為297 K,譜寬20 ppm,數據采集使用Topspin 3.2,核磁數據處理使用MestReNova 13.0。
2 結果與討論
2.1 對映體混合物核磁分析
(R,S)-4和(S,S)-4混合物核磁圖譜如圖2所示,根據化學位移值和化合物結構可以看出混合物核磁信號中,有三組對映氫核的化學位移值可供確定(±)-BINOL的ee值,這三組氫核分別為苯環氫(a)、芐基氫(b)和甲基氫(c),其在化合物結構中的位置見圖1標示,其化學位移值見表1。
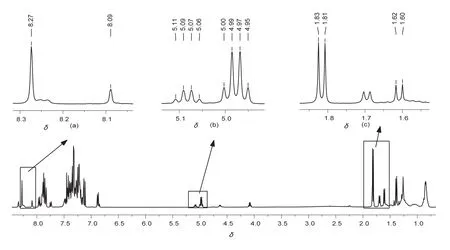
圖2 非對映體混合物400 MHz 1H NMR譜圖

表1 對映體混合物核磁共振化學位移
2.2 標準樣品數據處理
利用核磁法進行定量分析時可以采用峰高或峰面積,本實驗中采用峰高,可避免其它信號的干擾,相比峰面積結果更優。聯萘酚標準品共準備9份,ee值范圍為+80% - -80%,使用1.4小節中方法測試得到對映體混合物4的核磁數據,根據表1的位置標注化學位移,記錄下對應的峰高,根據三組對映氫核的信號分別計算(R)-BINOL的含量(R%),計算結果見表2。(R)-BINOL的含量(R%)及其ee值計算公式如下:

其中,HR為(R,S)-4結構中a、b、c位置處質子在1H NMR譜圖中的對應峰高,Hs為(S,S)-4結構中a’、b’、c’位置處質子在1H NMR譜圖中的對應峰高。
對映體混合物液相色譜譜圖數據處理時,也采用上述公式,只需將公式中的峰高替換為樣品對應的峰面積即可,計算結果一并列入表2。

表2 聯萘酚手性液相色譜方法與核磁共振方法(R)-BINOL含量和ee值測定結果
2.3 定量分析對映氫核的選擇
以標準品中(R)-BINOL的理論ee值和實際測量的百分含量作圖,考查其線性關系,如圖3所示,該圖采用苯環氫核磁信號計算樣品含量,R2為0.9999,具有很好的線性關系。液相色譜方法和芐基氫核磁信號兩者的R2均為0.9998,甲基氫R2為0.9988。

圖3 標準樣品理論ee值與(R)-BINOL測試含量標準曲線(苯環氫)
采用甲基氫信號計算樣品含量相對誤差比苯環氫和芐基氫要大很多,可能原因是甲基氫化學位移處信號復雜,基線難于處理平整,導致峰高計算誤差較大,因此不宜采用此處信號作為定量標準。芐基氫的核磁信號能很好地反映出標樣中(R)-BINOL的含量,結果與苯環氫相近,考慮到該處為四重峰,而苯環氫的核磁信號為單峰,便于數據處理,因此,優先選擇苯環氫信號計算樣品含量。
2.4 學生樣品測試
選取拆分前后的聯萘酚學生樣品,按照標樣測試方法進行測試,選取液相色譜和苯環氫信號進行數據處理,結果見表3。手性液相色譜方法測量ee值是目前公認的準確方法,因此測試結果以該方法所測得ee值為參照考查核磁方法的可靠性,由結果可知兩種方法有著較好的一致性。表3中學生1號樣品核磁結果為(R)-BINOL含量97.5%,此時(S)-BINOL含量為2.5%,其信號信噪比為SN= 10,可見若樣品中(R)-BINOL或(S)-BINOL含量大于97.5%時,核磁方法將無法提供準確的結果。

表3 聯萘酚學生樣品測試結果
2.5 相比手性液相色譜方法的優劣性
由前面實驗數據可以看出核磁方法測定聯萘酚對映體純度是一個切實可行的方法。相比液相色譜方法,核磁方法有以下兩個優點:一是測量速度快,一個樣品測試約需5 min,而聯萘酚手性液相色譜分析約需10 min,其他手性樣品可能需半小時以上;二是試劑消耗大大減少,液相色譜方法每測一個樣品會產生至少20 mL廢液,而核磁方法只需要消耗約3 mL試劑,大大降低了廢液的產生量。
手性液相色譜的關鍵主要在于手性色譜柱的選擇,目前商品化的手性柱能夠針對不同類別的化合物提供非常多的色譜柱選擇,而核磁技術測定對映體純度關鍵在于手性試劑的選擇,針對不同待測物需要尋找不同的手性試劑,相比液相色譜方法普適性要差,本文所用方法僅適合部分手性二醇和手性伯胺的ee值測定。
核磁共振方法因為信噪比的問題,其在檢測高純度物質時,難于檢測到微弱的雜質信號,因此該方法ee值測量結果有效性范圍為-95.0% - 95.0%。另外,在樣品前處理方面,核磁方法相對液相色譜方法更加繁瑣耗時。
在有機化學教學實驗“手性聯萘酚的制備”的樣品表征中引入核磁的方法進行樣品分析,讓學生可以和經典的液相色譜法進行效果比較,找出兩種方法的優劣性,從而拓寬其知識面。
3 結語
本文采用在聯萘酚樣品中添加2-甲酰基苯硼酸和(S)-(-)-1-苯乙胺的混合物作為手性試劑的方法,成功利用核磁共振氫譜技術測定了聯萘酚的對映體純度。在一定的ee值范圍內(-95.0% - 95.0%),與手性液相色譜方法相比,核磁方法具有很好的準確性且分析速度快。學生通過分析非對映體混合物復雜的核磁譜圖,可以進一步熟悉手性化合物結構并能深入了解核磁原理,能提升學生的基本科研素養。該方法適合作為有機化學教學實驗“手性聯萘酚的制備”的樣品表征手段。
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
兒童故事畫報(2019年5期)2019-05-26 14:26:14
中國生殖健康(2019年3期)2019-02-01 06:12:26
Coco薇(2016年2期)2016-03-22 02:42:52
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
海軍航空大學學報(2015年3期)2015-11-11 17:20:00
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
