盤點2021:近10 年世界新藥研發與藥品供求關系發展*
2022-03-21 02:58:24劉昌孝
中國藥業 2022年5期
關鍵詞:藥品
劉昌孝
(天津藥物研究院·釋藥技術與藥代動力學國家重點實驗室·天津市藥品監管科學研究會,天津 300462)
近兩年以來,新冠肺炎(COVID - 19)疫情暴發并持續蔓延,新冠疫苗和抗病毒療法受到了普遍關注,同時其他各疾病領域的藥物研發進展也并沒有絲毫放緩。隨著全球醫藥市場競爭愈加激烈,企業對新藥研發投入不斷增加,促進新的藥物靶點及治療方式不斷被發現和應用,創新藥研發將出現新的市場機會,因此各界對全球新藥研發進展的關注愈發強烈。為此,主要梳理了美國、歐盟和日本的新藥發展成績,介紹主要的突破性進展;從臨床失敗的案例分析以臨床需求為導向的新藥臨床研發的重要性;回顧仿制藥在世界醫藥格局中的地位,分析其為世界提供安全、有效、經濟、可及藥品及降低醫療成本的價值,還對處理藥物供需關系的影響因素進行了簡要分析。
1 2021 年創新藥物發展的主要成績
1.1 美國
根據年初Nature 雜志對2021 年美國食品和藥物管理局(FDA)批準新藥的盤點和FDA 網站公布的信息[1-4],年內批準了新分子實體(new molecular entities,NMEs)36 個,生物大分子藥物14 個,總計50 個,其中NMEs占72.00%,生物大分子藥物占28.00%。
筆者收集并統計了FDA 2011 年至2021 年批準的新藥數量(見表1),發現各年批準上市的NMEs 和生物大分子藥物的數量和占比情況基本一致。這11 年的數據顯示,NMEs 占主導地位(76.09%),生物大分子藥物僅占23.91%。FDA 的生物制品評估和研究中心(CBER)還批準了具有里程碑意義的mRNA 疫苗和CAR - T 細胞產品,但CBER 批準和應急使用授權(EUA)未被包括在年度新藥統計數據中。
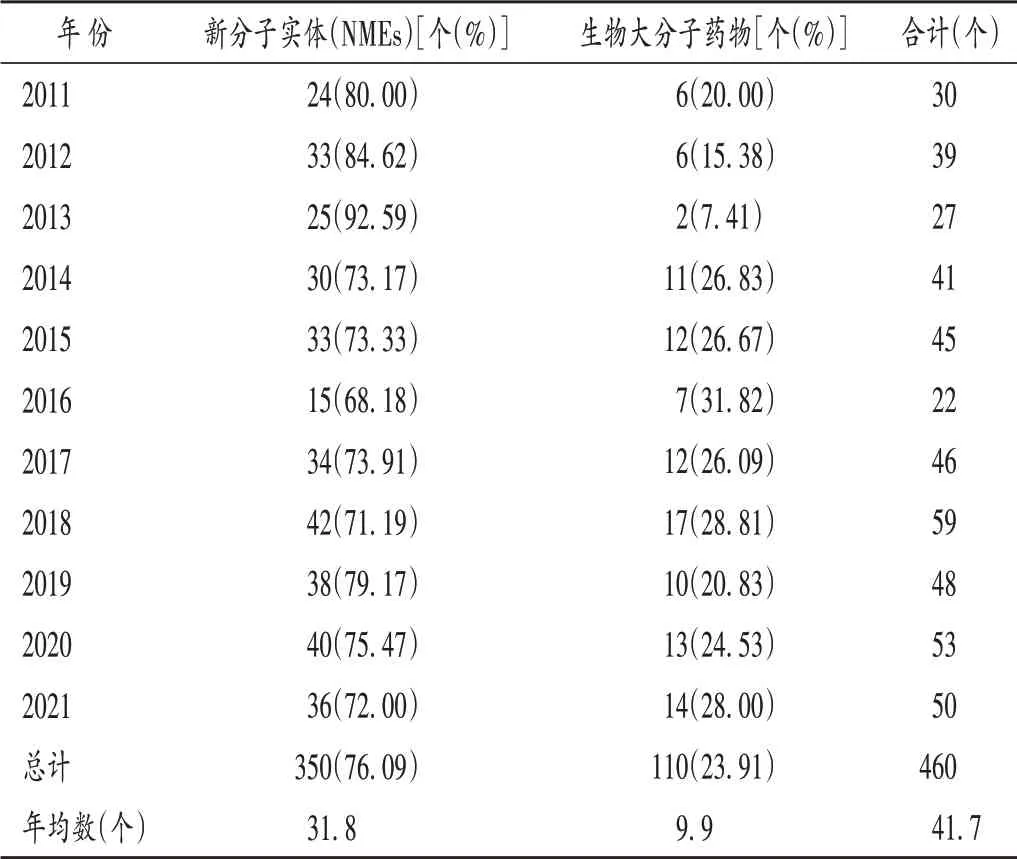
表1 2011年至2021年美國FDA批準上市新藥數量統計Tab.1 Number of new drugs approved by FDA from 2011 to 2021
生物藥方面,CBER 批準的疫苗是2021年“批準”的明星。尤其是輝瑞/BioNTech公司的COVID-19 mRNA疫苗開創了先例。BioNTech 公司于2020 年1 月開始研發,3 月與輝瑞公司達成合作,7 月進行Ⅱ/Ⅲ期臨床試驗,12 月便提供了應急使用授權(EUA)的安全性和有效性數據。在該計劃啟動僅1.5 年后,于2021 年8 月獲得FDA 完全批準,這已經超越一般疫苗發現和開發的平均時間10.7年的水平。
2021 年FDA 批準的10 個最引人注目的藥物,被認為是治療獲得新的突破,或是首個被批準用于某個適應證而滿足了醫療需求的藥物,或是同類藥物中首個被批準用于某個適應證。包括:百健公司的Aduhelm(aducanumab),輝瑞和BioNTech 公司的Comirnaty,輝瑞公司的Xalkori,Aurinia 公司的Lupkynis(voclosporin),默克公司的Tepmetko,基因泰克公司的Actemra,Chiesi Global Rare Diseases 公司的Ferriprox(deferiprone),艾伯維公司的Qulipta,諾和諾德公司的Wegovy,武田公司的Livtencity。2021 年新藥獲批的一大亮點是,借助FDA 快速通道認定和/或孤兒藥認定的比例高達80%,與2020年(占比68%)相比增加了12%。
癌癥治療獲得新的突破方面,值得介紹的是FDA批準安進(Amgen)公司的KRAS-G12C抑制劑sotorasib上市,這是癌癥治療藥物的一個里程碑。然而,盡管sotorasib 在突變非小細胞肺癌(NSCLC)患者中得到認可,但還沒有滿足期望,目前正在評估其與其他藥物聯合治療各種癌癥的療效。
FDA 2021 年批準的新藥中,默沙東公司first-inclass 缺氧誘導因子- 2α(HIF - 2α)抑制劑,用于治療與VHL(一種與血管浸潤性腫瘤相關的遺傳性疾病)相關腫瘤。對VHL疾病生物學數十年的研究表明,HIF-2α是氧感應的關鍵驅動因素,為新型抗血管生成藥物的發展開辟了道路。武田公司的小分子激酶抑制劑mobocertinib,能夠選擇性靶向exon 20 突變的表皮生長因子受體(EGFR)。FDA 批準的Livtencity,屬“孤兒藥”和突破性療法的NMEs,作為一種抗病毒藥物,其針對并抑制UL97 蛋白激酶及其天然底物。Livtencity 還獲得了歐盟的“孤兒藥”認定。除BMS 公司開發的用于多發性骨髓瘤的首款靶向B 細胞成熟抗原(B cell maturation antigen,BCMA,CD269)的CAR - T 細胞療法外,FDA 還批準了基于靶向CD19 的另外5 個CAR-T 細胞療法。BCMA 在B 細胞表面表達,已成為抗體、雙特異性藥物、ADC和細胞療法的試驗靶點。
葛蘭素史克公司的PD1 單抗(dostarlimab)是FDA批準的第100 個抗體藥物。其他5 個PD1/PDL1 抗體有望在2022年獲批準。基于檢查點抑制劑的研發,已成為“商業贊助商、臨床試驗和重復開發計劃的蜂擁而來”的促進藥物開發合作形式。強生公司獲批的新藥amivantamab,是靶向EGFR 和MET 的雙特異性抗體,用于EGFR exon 20 突變的NSCLC(這一類型癌癥對小分子EGFR 抑制劑具有耐藥性)。雙抗目前占臨床階段抗體治療的近20%。
FDA 還批準了兩款ADC 藥物,loncastuximab tesirine 是靶向CD19 的ADC,用于治療B 細胞淋巴瘤;Seagen/Genmab 公司的tisotumab vedotin 是靶向組織因子的宮頸癌ADC。2019 年,阿斯利康公司獲得FDA 批準的靶向人表皮生長因子受體2(HER2)的ADC trastuzumab deruxtecan 的臨床數據表明,與抗體和之前批準的ADC相比,新型ADC具有更好的療效。
1.2 歐盟
截至2021年12月17日,歐洲藥品管理局(EMA)已批準49 個新藥(新活性物質,new active substance),包括全球首批上市的新藥有7 個。其中,小分子藥物32 個(65.31%),單克隆抗體藥物13 個(26.53%),新冠疫苗2 個(4.08%),細胞和基因療法(cell and gene therapy,CGT)2個(4.08%)[5]。
從企業新藥獲批上市數量來看,羅氏制藥成為大贏家,有5個新藥獲批上市(包括與其他藥企聯合開發,下同),數量最多,其次是阿斯利康公司(4 個)、強生公司(2個)、百時美施貴寶公司(2個)、再生元公司(2個)、拜耳(1個)、GSK公司(1個)等。
全球首批上市的7個新藥包括:1)jemperli為PD-1阻斷抗體,是全球首個用于復發或晚期子宮內膜癌的藥物。2)tralokinumab 用于治療中度至重度特應性皮炎的成人患者,是全球首個獲批治療特應性皮炎的單克隆抗體療法。奧維昔巴特(Odevixibat)用于治療膽汁酸在肝臟中積聚,該藥被歐盟指定為孤兒藥。3)elivaldogene autotemcel 是孤兒藥,用于治療18歲以下早期腦細胞白質營養不良(CALD)兒童患者。4)伏索利肽(Vosoritide)也是孤兒藥,用于治療2 歲至生長板閉合的兒童軟骨發育不全癥(achondroplasia)。該病是一種罕見的遺傳性疾病,由成纖維細胞生長因子受體3(FGFR3)基因突變引起,這種突變會影響身體幾乎所有骨骼的生長,導致身材非常矮小。5)Voxzogo 是全球首個獲批用于治療軟骨發育不全兒童的藥物,在歐盟和美國均被指定為孤兒藥。6)bimekizumab,用于治療適合系統治療的中度至重度斑塊型銀屑病成人患者。Bimzelx 是全球首個獲批的旨在同時選擇性抑制IL - 17A 和IL - 17F 的斑塊型銀屑病的治療藥物。7)regdanvimab,用于治療不需要吸氧且疾病可能發生進展的成人COVID - 19患者。
1.3 日本
截至2021年11月30日,日本獨立行政法人醫藥品醫療器械綜合機構(PMDA)全年共批準52個新藥上市,包括新活性成分(new active ingredient)、疫苗、血液制品等,其中全球首次批準的新藥有7 個(13.46%),20 個(38.46%)新藥被授予孤兒藥資格[6-7]。
獲批藥物適應證方面,抗腫瘤藥物數量很大,共有15 個 新 藥(28.85%),激 素 類、代 謝 病 藥 物8 個(15.38%),心血管系統疾病、帕金森病、老年癡呆癥藥物5 個(9.62%),呼吸系統藥物、抗過敏藥物、感覺器官藥物5 個(9.62%),胃腸道用藥、皮膚科用藥、免疫抑制劑等藥物5 個(9.62%),疫苗和抗毒素血清等藥物4 個(7.69%),抗感染藥物3 個(5.77%),泌尿生殖系統藥物、中樞/ 外周神經系統藥物、血液制品各2 個(3.85%),泌尿生殖系統藥物1個(1.92%)。
值得一提的是,日本對抗感染藥物的重視。三聯復方制劑Recarbrio(relebactam hydrate/ imipenem hydrate/cilastatin sodium),用于治療對Recarbrio敏感的大腸桿菌、檸檬酸桿菌、克雷伯菌、腸球菌、沙雷菌、銅綠假單胞菌及不動桿菌屬(限于對碳青霉烯類抗菌藥物耐藥的菌株)感染的患者。二聯復方制劑Ronapreve(casirivimab/ Imdevimab)在全球范圍內首次獲批,用于治療SARS - CoV - 2 感染引起的疾病。單抗藥物Xevudy(sotrovimab)用于治療SARS - CoV - 2 感染引起的疾病。
從審批形式來看,獲批的罕見病藥物有21個,除因疫情需求特例批準6 個(均為COVID - 19 治療藥物)、優先審批1 個新藥(Astellas Pharma 公司開發的ADC)外,其他新藥均是普通審評形式獲批。這說明日本對采用優先審批程序是嚴格把關的。
2 仿制藥的世界地位并未動搖
2.1 仿制藥發展是世界合理藥物結構的需要
世界需要合理的藥物結構滿足醫療衛生需求,各國的藥品結構是事關國家安全和國家主權的戰略問題之一,其仿制藥不僅是保障社會穩定、防病治病、疫情需求、戰備需求、經濟發展、人民生活和生命質量的民生問題,更是關系國家、政府和企業安全的政治問題。雖然創新藥是體現國家創新能力的主題,但從世界“創新藥-仿制藥-非處方藥-特殊藥”結構格局來看,仿制藥仍是世界醫療衛生需求的主體。
全球仿制藥行業發展歷史悠久。1984 年美國Waxman-Hatch 法案(也稱《藥品價格競爭與專利期補償法》)獲得通過,仿制藥的研發和申請流程被簡化。美國據此成為仿制藥的受益者,成為全球真正的仿制藥大國,其仿制藥占全球40% 的市場份額。根據該法案,仿制藥只需要提供簡化新藥申請(ANDA),以生物等效性試驗和文獻數據來代替安全有效性試驗,開發成本大幅降低。同時,該法案還規定了“Bolar 例外條款”,允許仿制藥廠家在原研專利期內開展研發、申報和生物等效性試驗,鼓勵其挑戰原研的專利,對首仿藥給予180 天的市場獨占期。該法案的實施,一方面保護了原研廠家的利益,另一方面也為仿制藥行業的發展鋪平了道路。此后,全球仿制藥市場開始蓬勃發展。有數據顯示,近年來全球仿制藥獲批數量不斷增長,2020 年因疫情影響有所下降;美國FDA 共批準909 個仿制藥,其中737 個為完全批準,172 個為臨時批準。同時,全球仿制藥市場規模也在不斷增長,根據東方財富網2021 年9 月21 日公布的數據,2020 年全球仿制藥市場規模達4 520億美元,預計2021年接近5 000億美元。
美國國內醫療使用藥品的95% 也是仿制藥,其中86% 以上的處方藥是仿制藥。美國FDA 仿制藥年報顯示,2013年至2020年8月間節省醫藥費用2.2萬億美元,2017年至2020 年每年節省醫藥費用2 500~2 900 億美元。筆者統計發現,2013年至2021年美國企業每年申請仿制藥約1 000 個,獲得批準和暫時批準的仿制藥約800 個(表2)。FDA 暫時批準,意味著因專利權、獨占權因素,等待專利過后即可在美國上市。部分FDA 暫時批準的藥品符合要求(在美國上市的FDA 質量、安全和有效性標準)后可正式獲得批準。
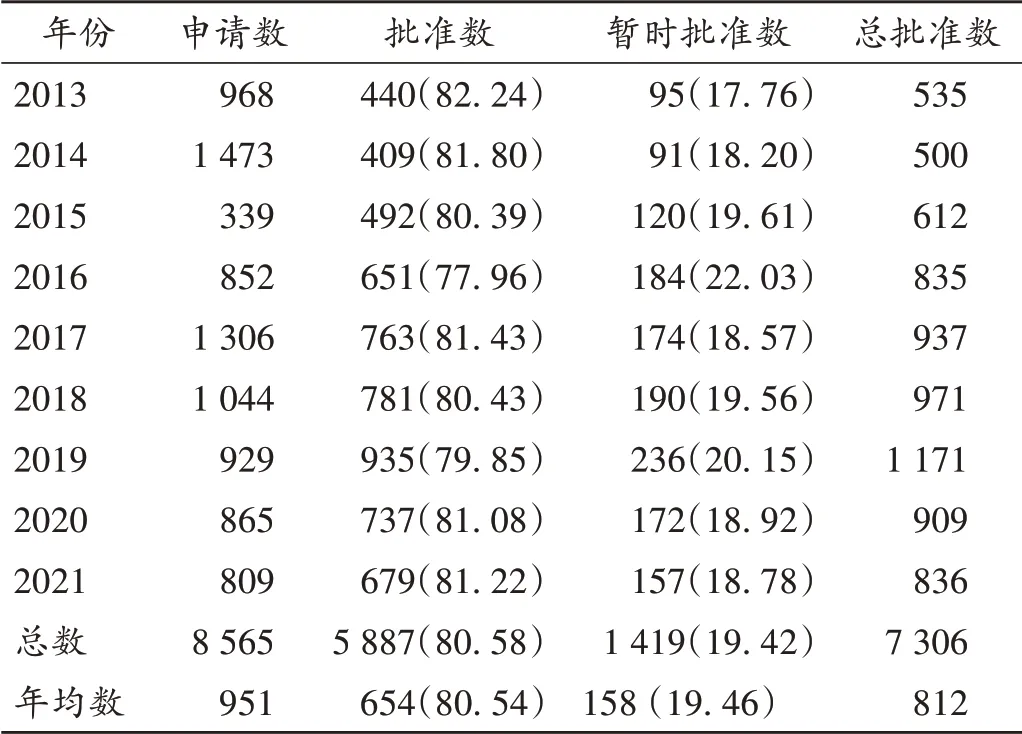
表2 2013年至2021年美國仿制藥申請與批準數量統計[個(%)]Tab.2 Number of generic drug applications and approvals in the USA from 2013 to 2021[n(%)]
2.2 2021 年全球仿制藥排名前15(TOP15)的企業
據國際政府基準測試協會(IGBA)2020 年11 月發布的數據顯示,仿制藥滲透率排名前5的國家分別是印度(97%)、美國(92%)、約旦(85%)、澳大利亞(84%)、日本(77%)。仿制藥具有高性價比特點。仿制藥做得好,需要精湛的制藥技術、堅持不懈的改良來保證質量,同時還要配合高超的銷售技巧才能賣得好。據IMARC 集團的最新報告,全球仿制藥市場規模2020年達到3 860億美元,2026 年預計將達5 170 億美元,2021 年至2026 年的復合年增長率將達4.9%。2020 年,全球仿制藥排名前15(TOP15)的銷量達727 億美元。根據中國前瞻產業研究院等總結的2021 年全球TOP15 的仿制藥企業 中[8-11],印度有7 家,占了近50%。據成招榮[8]的2021 年全球仿制藥行業市場現狀與區域市場分析顯示,印度是名副其實的仿制藥大國,仿制藥滲透率最高(97%),居世界第一。
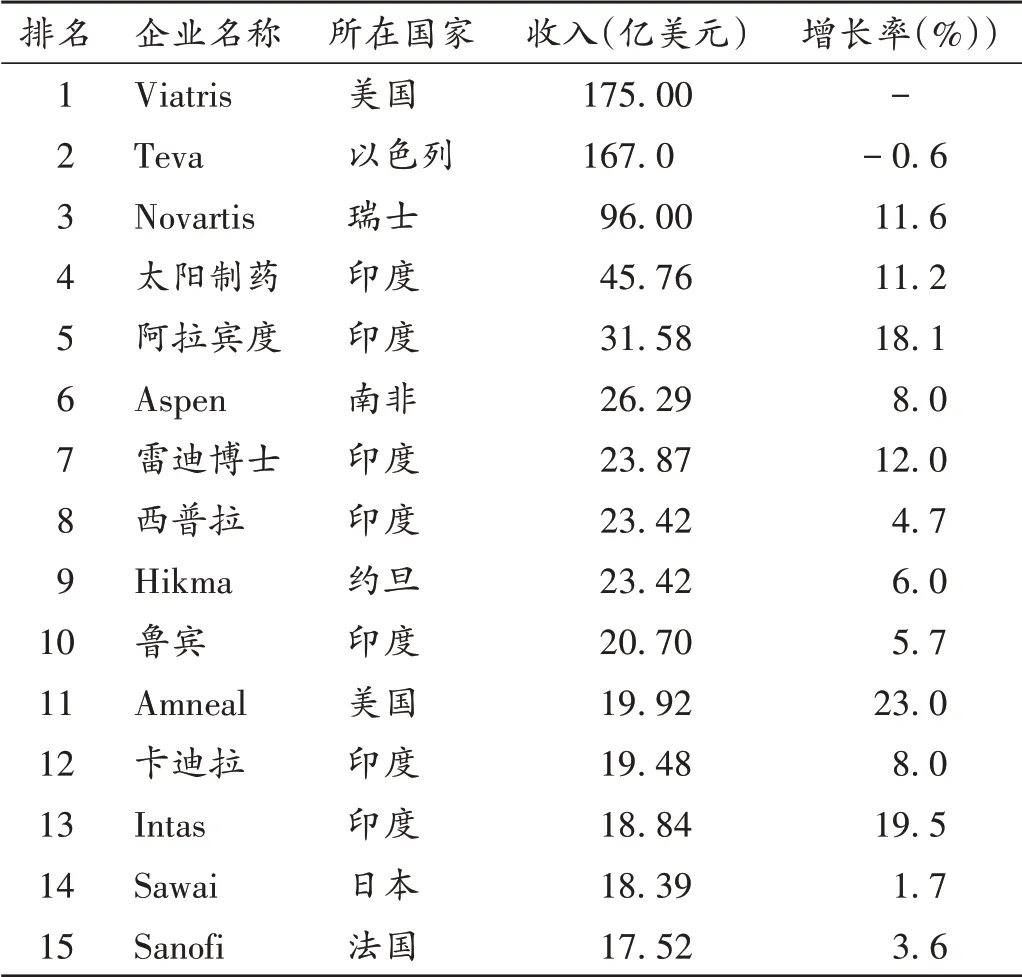
表3 2021年世界排名前15(TOP15)的仿制藥企業統計Tab.3 The world's top 15 generic drug enterprises in 2021
值得注意的是,新成立的Viatris 公司(NASDAQ:VTRS)的發展態勢。Viatris 公司是2020 年11 月成立的新型醫療保健公司(Mylan 和輝瑞的Upjohn),匯集了科學、制造和分銷專業知識,具有經證明的監管、醫療和商業能力,為超過165個國家和地區的患者提供高質量藥物。其產品組合包括跨越非傳染性疾病和傳染病等廣泛治療領域的1 400 多個經批準的分子藥物,包括全球公認的品牌藥、復雜的仿制藥和品牌藥、越來越多的生物仿制藥,以及各種非處方消費品。
日本2002 年開始在醫療機構推動使用仿制藥,引進激勵機制,激勵醫師、藥房和患者使用。仿制藥替代率2017 年超65%,2018 年達80%。日本仿制藥企業的集中度也顯著提高,1975 年近1 000 家,到2016 年集中到近200 家(含OTC 制藥)。日本排名前3 的仿制藥企業(Sawai,Nichi - Iko,Towa)分別生產1 028 個、765 個和738 個仿制藥品種,共計2 531 個品種。企業重視仿制藥的研發投入,研發費用占比最高達到銷售額的9%。政府也鼓勵仿制藥向外擴展國際市場。
印度被譽為“世界藥房”,仿制藥是其制藥業最重要的組成部分,已成為全球仿制藥行業的主要貢獻者之一,以97%的仿制藥滲透率排名世界第一。目前,擁有60 個治療類別的60 000 個仿制藥品牌,是全球最大的仿制藥供應商,占全球供應量的20%。全球排名前15的仿制藥企業中,印度就占7 家,其排名前6 的企業均以仿制藥為主體,生產2 000 多億片制劑銷往全球[8-10]。
以色列Teva(梯瓦)公司是全球最大的仿制藥公司,擁有5 萬多名員工,2017 年銷售額224 億美元,列全球制藥企業銷售額排行榜第11 位。梯瓦公司的發展模式整體上非常成功,非常值得研究和學習。梯瓦公司利用Waxman-Hatch 法案,通過挑戰專利、打破仿制藥競爭壓力、提高自身產品覆蓋范圍來增加公司的產品數量。
3 臨床需求與快速通道審批的新藥
3.1 美國快速通道審批新藥的發展
美國FDA 于1988 年引入快速通道(fast track)機制,在收到制藥企業的主動申請后60天內給出答復;對于進入快速通道的藥物,將進行早期介入、提前指導以達到少走彎路、加快整個研發過程的效果;制藥企業還可分階段遞交申報資料,而不需要一次性遞交全部材料。到2014 年,美國FDA 提供了4 條特別審批通道,即快速通道(fast track)、優先審評(piority review)、加速批準(accelerated approval)和突破性藥物(breakthrough therapies),以適應特別需求的品種獲得早日上市的機會[11-14]。
為了促進罕見病(rare diseases and/ or conditions)治療藥物的研發,美國早在1983 年就通過了世界上首個孤兒藥法案(Orphan Drug Act,ODA)。ODA 自頒布以來取得了巨大成功,已成為制藥行業、患者和政治家的共識,且成功帶動了其他國家和地區的相關立法和實施。從一定程度來看,孤兒藥審批可以說是一種快速通道審批的途徑。ODA 界定了罕見病及孤兒藥,對罕見病的界定融入了經濟與社會因素,其頒布與修訂涉及眾多利益相關方,包括聯邦政府、企業、患者,民眾和媒體發揮了重大推動作用[15-19]。該法案雖然豐富了孤兒藥市場,但也造成部分藥品的高價問題。
3.2 2021 年通過FDA 快速通道獲批的新藥
全球藥政法規事務咨詢公司奧來恩在題為“2021年FDA CDER 批準新藥的分析-創新藥獲得FDA 批準的加快通道”中的分析顯示,2021年獲批的50個新藥有5 種情況,包括:1)快速通道認定(fast track designation,FTD)有18個(36.00%),有助于加快新藥研發及申報資料審評進程;2)突破性治療認定(breakthrough therapy designation,BTD)有14個(28.00%),享有快速通道認定的所有優惠政策,可獲得與FDA 的溝通交流與技術指導,從而加快藥物的后續研發;3)優先審評認定(priority review designation,PRD)有34 個(68.00%),僅需CDER確定某個藥物獲批后將顯著改善某種嚴重疾病的治療有效性和/或安全性;4)加速批準(accelerated approval,AA)有14 個(28.00%),對于一些治療嚴重疾病且與現有藥物相比具備顯著優勢的藥物,FDA 可以根據合適的能夠預測臨床受益的替代終點來批準藥物上市,有利于把那些可以提供重大醫療進展的藥物更快帶給市場;5)孤兒藥認定(orphan drug designation,ODD)有26個(52.00%),這是對美國患病人數少于20 萬的疾病治療藥物的一種特殊審批形式。但是,目前用于治療罕見病的新藥仍非常有限,僅有很少或沒有可用的藥物。
3.3 對藥品快速審批的質疑
2015 年夏天,美國政府又提交了《21 世紀治愈法案》(21st Century Cures Act),旨在進一步推動藥物審批加速。但一些最新的研究認為,加速審批或許會使一些無效甚至是有害的藥物進入市場。
阿爾茨海默病(Alzheimer's disease,AD)是一種進展緩慢的神經退行性疾病。100多年來,全球只有5個用于臨床治療的藥物,且臨床獲益并不明顯。過去20多年里,相繼320 余個進入臨床研究的藥物均宣告失敗。過去幾十年里,幾乎所有靶向β-淀粉樣蛋白的治療策略均在臨床試驗中“全軍覆沒”。
2021 年6月7日,美國FDA采用加速審批渠道批準了自2003 年來首個AD 新藥aducanumab(Aduhelm)。這一藥物是AD治療歷程中近20年來的“超級重磅”新藥,但也引發了對FDA 藥物審批標準的重大爭議[20]。FDA的這一決定已經導致了3 名FDA 神經系統藥物咨詢委員會專家辭職,包括Joel Perlmutter(華盛頓大學)、David Knopman(梅奧醫學中心)和Aaron Kesselhein(哈佛大學)。Aaron Kesselhein 甚至稱這是美國近期最糟糕的藥物批準決定。3 名專家曾聯合在JAMA 上發表觀點性文章,對aducanumab 進行評估[21]。接受高劑量aducanumab 治療組患者,衡量認知能力的CDR - SB 評分在EMERGE 臨床試驗中降低22%(評分降低意味著疾病癥狀惡化速度減緩),在ENGAGE 臨床試驗中反而升高了2%[22]。
aducanumab獲批引發爭議的原因有2個,一是兩項關鍵Ⅲ期臨床試驗結果不一致,二是目前學者們對解決了β-淀粉樣蛋白的沉積就能治愈AD這一觀點存在質疑。研究者試圖尋找治療AD 的特效藥物,然而這些研究的療法雖能成功降低了患者的Aβ 水平或消除了大腦中的β-淀粉樣蛋白沉積,但對患者認知能力衰退卻沒有幫助。aducanumab是基于β-淀粉樣蛋白假說而開發研制的一種單克隆抗體藥物,約有35%的高劑量治療患者會出現藥物相關的腦部水腫等反應[23]。基于療效的不確定性和潛在的安全隱患,外圍專家咨詢委員會一致不同意獲批aducanumab。2020 年10 月,有2家公司向FDA 尋求對該藥物的監管批準,但能否獲批仍存在很大的不確定性。
3.4 以患者為中心、以臨床需求為導向的新藥研發挑戰
如何適應當前新藥審批中以患者為中心、以臨床需求為導向新要求的挑戰是新藥研發面臨的難題。當臨床試驗出現陰性臨床結果,就意味著新藥的特點未滿足臨床需求,肯定是失敗的重要原因。表4 總結了2021年最終臨床失敗的10個創新藥[24-34],并分析了失敗原因。

表4 2021年最終臨床失敗的10個創新藥Tab.4 Ten innovative drugs with final clinical failure in 2021
4 不同國情的采購模式影響藥品供求關系的發展
4.1 醫藥產品供求關系的復雜性
作為特殊產品,基本屬性決定了醫藥產品發展是一項系統工程,受多方因素影響,也受不同國情所形成的特殊監管模式的影響,而呈現出復雜的供求關系(圖1):以投資、制造、商業為第一供給方,醫療機構為主動消費者和第二供給方,患者為被動消費者(醫師開什么藥就用什么藥)和健康受益者(用對了藥時患者康復,用不對藥患者不但得不到康復,甚至出現生命安全問題和經濟損失),在上述基礎上形成社會效益(以安全、有效、經濟效益為特征),不同國家政府出臺的政策明顯影響著四方關系。醫藥產品供求關系不僅與國家發展政策、鼓勵創新和適度發展仿制藥提高藥物供給體系及格局有關,還與藥品采購政策、醫療保障能力及商業保險能力有關。因此,如何平衡四方關系顯得十分重要。

圖1 醫藥產品供求關系的復雜性Fig.1 Complexity of supply-demand relationship of pharmaceutical products
1999 年,WHO 在日內瓦頒布《WHO 良好藥品采購準則》(operational principles for good pharmaceutical procurement)[35],明確提出應采購最具成本- 效益的藥品,選擇可靠的供應商購買優質藥品,保證產品及時交付,并且盡可能降低采購總成本。通過對美國、英國、德國、荷蘭4 種藥品采購模式的特點和影響進行分析,筆者認為,藥品采購政策是各個國家特定國情下的產物,國際上并沒有統一的采購模式可以完全適用于所有國家。各國采購模式的共性之處在于,降低藥品價格、節省醫保基金支出是藥品采購的核心出發點,價格是主要但非唯一的考量因素,綜合性價比越來越受到重視。當過度關注價格時,應考慮可能產生的供應問題,以及品種遴選、頻繁采購對患者用藥的影響。
4.2 美國的藥品采購模式
美國是實行醫藥分開政策的國家,集團采購組織(group purchasing organization,GPO)主要針對各類醫療機構住院用藥進行采購。1910 年以后的數十年,隨著醫療費用的急劇上漲及保險償付比例的下降,醫院成本壓力越來越大,促進了美國GPO 的發展。GPO與藥品生產商或經銷商談判獲得價格折扣,各個GPO 在醫院業務上也相互競爭[36-37]。GPO 運行費用主要依靠生產商提供的合同管理費用(contract administration fees,CAFs)。1987 年,美國國會在反回扣法(The Anti-Kickback Statute)除外條款中添加“GPO 安全港(safe harbor)”條款,允許GPO 向供應商收取不超過購買價格3%的合同管理費用[38]。
美國GPO 采購流程包括發標、評標、談判及磋商、訂立合同、供應商交付等。在發標方面,GPO 會對醫療機構開展調研并收集其采購需求,在網上發布招標方案,要求供應商投標。在評標方面,供應商的選擇主要由醫療機構的醫師、藥師和其他臨床專家組成的評審委員會決定;要考量非財務因素,如供應商供應保障能力、服務和信譽水平、產品質量和安全、產品原料藥來源、美國FDA 監管警告記錄等。醫療機構確定供應商后,由GPO 與供應商談判磋商合同細節,形成會員醫院統一適用的合同條款。對醫療機構而言,并沒有被強制要求必須使用GPO 制定的合同,所有合同簽訂都是自愿的,也經常存在醫療機構、療養院等不使用GPO 合同的情況。通常只有在獲得更大折扣時,GPO 才會簽訂單一貨源合同[39]。
目前,GPO 在美國醫藥供應鏈中發揮著越來越重要的作用,其最大優勢就是節約成本。GPO 每年為醫療保健系統節省費用550 億美元,10 年來合計達8 640 億美元[40]。對供應商而言,參與GPO采購可以節省交易成本、形成規模效應,并且通過提前備貨來優化庫存管理,但可能導致產品市場競爭程度下降、用藥短缺等問題[41-42]。另外,也存在關于對GPO 可能產生壟斷、限制競爭、影響產業創新的質疑[43]。
4.3 英國的藥品政府采購機制
英國國民醫療衛生服務體系(National Health Service,NHS)始建于1948 年,是基于財政預算對所屬醫療機構實行高度計劃性管理的衛生服務體系[38]。英國藥品政府采購是指“針對公立醫院使用的藥品,包括品牌藥、血液制品、生物類似物、仿制藥,英國衛生部商業藥品處(Commercial Medicines Unit,CMU)均可組織采購”[43-44]。
就仿制藥采購而言,在采購主體上,由CMU 代表衛生部和NHS 統一管理醫院藥品采購、供應等工作,同時藥品采購工作得到國家藥品供應小組、藥品市場供應小組等部門支持。CMU 會充分考慮藥品的特點、使用場景、用藥安全風險。凡是一家企業連續2次中標,即使第3次報價最低,也不能中標,從而避免壟斷、保證市場供應。在中標結果執行上,醫院按照框架協議和供應商簽訂實際購買合同,理論上不允許醫院與供應商進行二次議價,實際上醫院出于成本考慮,仍會與供應商談判,壓低采購價格。
英國非常重視藥品政府采購在藥品價格調控中的作用。英國公立醫院藥品采購僅占NHS 藥品費用支出的20%~30%,其中仿制藥采購數量占60%,金額占20%[43,37]。英國政府主導藥品集中采購,在一定程度上能起到促進競爭、降低采購價格、提高NHS 資金使用效率的作用。
5 未來5 年的展望
5.1 全球醫藥創新發展的基本格局不會明顯改變
分析全球醫藥創新的未來形勢,可能呈現六大趨勢:1)全球藥品市場增速加快,特別是疫情需求的影響越來越明顯。2022 年需求預計將達11 000 億美元,年增長8.4%。2)在美國的帶動下,全球加速審批的新藥占比越來越高。3)制藥巨頭企業持續加碼研發投入,而回報率越來越低(表5),可能出現世界創新研發和仿制藥產業向亞太遷移。4)多元化研發模式將迎來國際合作形式的變化,由于國際、國家、區域利益的關系,受大型制藥企業研發效率不高的影響,外資制藥企業紛紛關閉或出售在某些國家研發中心,傳統研發模式開始變革,轉而與研發效率更高的早期研發型小企業合作。5)雖然生物仿制藥研發方興未艾,但由于研發成本高、品種多、應用范圍小、療效有限、市場承受度等問題,未來5 年獲批生物大分子藥物的數量超過NMEs(占70%)的可能性較小。孤兒藥研發活躍,但上市后拓展新的適應證有難度,最近很多重磅交易都涉及到孤兒藥,其新藥研發將會因品種差異帶來的利好也有所不同。6)仿制藥是各國滿足臨床需求的基本來源,即使美國用20%的GDP 用于醫療衛生開支,也需要仿制藥來節省數以千億美元計(每年約2 600 億美元)的藥費開支,仿制藥因低價、有效、安全特點仍是發展中國家滿足民生需求的主力。
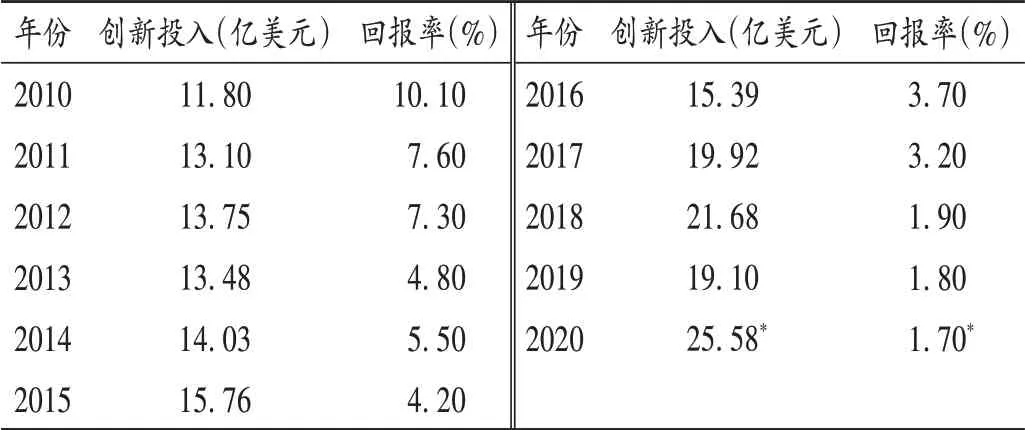
表5 創新投入居高不下與回報率逐年降低Tab.5 Continuing high investment in innovation and decreasing rate of return year by year
從歷年新藥研發投入來看,臨床費用是開發研究的大頭,其中臨床前研究僅占30%(藥物篩選5%,醫學研究10%,藥物評價研究15%),隨著臨床試驗的開展,一期比一期高(臨床Ⅰ期5%,臨床Ⅱ期15%,臨床Ⅲ期50%),臨床研究耗時也在6~8 年不等。因此,不僅需要重視臨床前開發計劃的先進性、生物學模型精準、藥靶與臨床疾病的關聯性,更需要把握好臨床進展的每一關。
新藥研發的大多數項目會以失敗告終,現在進入Ⅰ期臨床試驗的藥物90%以上不會上市。現在的研發項目幾乎沒有可能100%“去風險”,做一個準一個不現實,失敗貫穿于整個研發過程中。“去風險”的具體執行就是更加個性化,每個公司各有“套路”,對風險的忍受程度也各有不同。同時,研發決策質量與市場、監管、支付環境變化也有關聯,還需關注靶點組織的藥物濃度、藥物與靶點是否以預期方式結合、相關藥理變化等。合理設計、高效執行Ⅱ期和Ⅲ期臨床試驗是研發的關鍵。
5.2 必須重視研發路徑的創新發展
針對創新藥物研發周期長、投資高、風險高、成功率低和回報率低的“一長兩高兩低”特點,有一些跨國制藥企業對已形成近60年的研發模式和路徑進行了大量革命性工作,如“加快審評”“孤兒藥政策”“靶點驗證”“生物標志物識別”“臨床終點”“真實世界研究”等,取得了一定成效,目前已經有很成熟的化合物研發管道(pipeline)。新藥研發是一個厚積薄發的過程,在傳統的研發體系下,沒有pipeline 的經驗累積是新的原研藥企業遇到的很大障礙,為改變此局面,人工智能(artificial intelligence,AI)被寄予厚望。但新藥研發如此復雜,AI仍不能完全代替人類大腦,在理解生物學各個環節的局限性也導致了新藥研究的很多問題難以解決。
AI 應用于醫藥研發更重要的是落地場景的選擇,將科技和傳統醫藥結合起來。AI 可以應用于鎖定致病蛋白質、篩選對蛋白質起作用的藥物成分、評估藥物成分的安全性并決定合成方法、制訂臨床試驗計劃等,目前在用于構建新型藥物分子、藥物挖掘、篩選生物標志物、新藥有效性/安全性預測、精準醫療和新型藥物靶點與組合療法等方面已有所進展。
BROWN 等[45]對2010 年至2019 年間美國FDA 批準的378 個新藥和27 個生物仿制藥,按年份、治療領域、模式、給藥途徑、一流指定、批準時間和快速審查類別的批準號進行了分析和評估,結果顯示,腫瘤學仍然是最大的治療領域(25%),其次是感染(15%)和中樞神經系統疾病(11%);監管激勵措施是有效的,孤兒藥和根據GAIN 法案批準的抗菌藥物增加就可以證明;臨床開發時間可能正在增加,也許是孤兒藥適應證增加的結果;小分子藥物繼續且主要遵守“5法則”(Ro5)參數,但隨著反義寡核苷酸(ASO)、小干擾RNA(siRNA)和抗體導向偶聯物(ADC)的批準,新模式的創新正在迅速發展。最后,他們還詳細討論了解決為滿足臨床需求領域的新靶點和科學突破。
表型藥物發現(PDD)直接使用生物系統進行新藥篩選,雖然已被證明在發現具有新機制的藥物方面是有效的,但為了更廣泛應用,需要解決關鍵挑戰:由于缺乏有效的工具來處理和優先處理命中,合格的潛在客戶和過載的管道的進展,以及具有不良機制的線索的進步。基于人的表型平臺正在應用于整個發現過程,用于命中分類和優先級排序,消除具有不合適機制的命中,以及通過基于途徑的決策框架支持臨床策略。隨著這些方法使用的增加,將獲得推動更好決策的能力,從而促進PDD 更大范圍的采用[46]。BLOOM等[47]提出的構建藥物發現模式,也提供了有益的探索方向。
RAYNER 等[48]認為,利用最新技術,對于未來抗感染藥物的模型知情(model - Informed)藥物開發很重要。KASHTE 等[49]認為,COVID - 19 疫苗研發的快速發展、影響因素、挑戰和未來前景必須引起重視。
新藥發現和開發成本高昂、耗時且往往效率低下,在研發過程中會出現許多失敗。在AI的支持下,語言模型(LM)改變了自然語言處理(NLP)的格局,為更有效地改變治療開發模式提供了可能。LIU 等[50]總結了AI驅動的語言模型進步及其在幫助藥物發現和開發方面的潛力。在開發抗新型冠狀病毒策略、新療法的潛在作用,包括藥物再利用、可能導致大流行的傳染病研究方面,特別強調了AI驅動的語言模型在目標識別、臨床設計、監管決策和藥物警戒方面的機會[51]。
在新藥研發復雜而漫長的過程中,投資者必定要引入商業市場,而商業市場需要服從臨床需求才能實現。研發過程通常跨越數年,且由于高損耗率而產生巨大投資成本,迫切需要使用AI等創新技術來改進。不同AI工具正在被應用來支持藥物開發過程的4個步驟,即藥物發現的基礎研究、臨床前階段、臨床階段和上市后臨床研究。GALLEGO 等[51]綜述認為,AI 已被證明有用的一些主要任務包括識別分子靶標、尋找命中和先導化合物、合成類藥物化合物以及預測ADME-Tox,這篇綜述一方面使藥物開發中使用的一些關鍵AI方法的數學視野更接近藥物化學家,另一方面使藥物開發過程和不同模型的使用更接近數學家。JIMéNEZ - LUNA等[52]也就藥物發現中AI 問題的最新進展和未來展望進行了分析。
5.3 必須重視臨床研發路徑的革命
根據世界新藥研發的現實情況,筆者認為,“必須重視臨床研發路徑的革命”且十分關鍵。其理由是:1)在整個研發過程中,70%的成本消耗于臨床試驗;2)各種快速審評審批程序對臨床前評價發揮的作用并不重要;3)快速審批通道為提前上市開了“綠燈”,但留下的安全有效“尾巴”成為政府監管和企業責任處理的難點;4)從大量Ⅱ期和Ⅲ期臨床試驗失敗的案例可見,影響臨床安全有效的問題還很復雜,以患者為中心、以臨床需求為導向的新理念是新藥研發的挑戰和難題。
近年來,為了適應免疫療法、再生醫學、細胞和基因療法等創新產品,全球藥物開發格局發生了巨大變化。全球COVID - 19 疫情大流行還導致負責批準新藥的主管部門實施與之相適應的監管途徑,以便及早獲得創新療法和疫苗,但面臨著開發時間短,或患者人數少,或藥品功能需要完整的新生產等問題的新挑戰[53]。
臨床藥理學是對人類藥物的研究,包括從首次人體研究到隨機對照試驗(RCT)和大人群的收益風險比評估。無論未來發展如何,開始人體藥理學研究之前,關于藥物作用機制和嚴格研究設計的可靠的初步數據仍將至關重要。盡管替代設計(務實試驗、平臺試驗等)正在出現,目前隨機對照試驗仍是評估人類藥物療效的黃金標準。AI、機器學習和基于互聯網的試驗等新技術的貢獻,有可能改進藥物開發。在精準醫學領域,新的疾病表型和類型可能有助于確定新的藥理學靶點、治療反應者和有藥物不良事件風險的患者。在這樣一個不斷變化的環境中,通過轉化研究,透明地共享臨床試驗數據,以及加強藥物專家、患者和公眾之間的互動是優先行動領域[54]。轉化科學的使命是為改善人類健康的干預措施的開發和傳播帶來可預測性和效率。許多轉化研究中心成立,在廣泛的轉化領域,體現、實施和支持取得了實質性進展,從診斷和藥物開發到臨床試驗,從實施科學到教育等方面對未來創新研究正發揮著作用[55]。
美國FDA 一直積極推動在藥物開發中使用真實世界數據(RWD)。RWD 可以產生重要的真實世界證據,反映使用治療的真實世界臨床環境。與此同時,AI(包括機器和深度學習,ML / DL)方法越來越多地用于藥物開發過程的許多階段。AI 的進步也為分析大型多維RWD 提供了新的策略。因此,有學者對過去20 年的文章進行了快速評估,以概述同時使用AI和RWD 的藥物開發研究,發現最受歡迎的應用是不良事件檢測、試驗招募和藥物再利用,同時還討論了當前的研究差距和未來的機會[56]。
5.4 需要從國際戰略高度認識仿制藥發展
2021 年2 月11 日,美國FDA 藥品評估與研究中心(CDER)的仿制藥辦公室(OGD)發布了2020年度報告,該報告是OGD 第6 份年度報告。OGD 認為,提供更多可負擔(經濟可及)的藥物仍然是FDA 的公共衛生重點,仿制藥的競爭可以降低藥品價格,并改善美國患者的藥物可及性。該報告指出,目前美國每10張處方中就有9 張使用仿制藥。在過去的近10 年中,仿制藥為醫療系統節省了2.2 萬億美元[57]。COVID-19 疫情期間,OGD迅速將重點放在優先考慮與COVID-19相關的仿制藥品的申請評估。OGD 和整個仿制藥計劃還保持了FDA的嚴格標準,用于基于質量數據和可靠的科學依據評估非COVID-19產品。
OGD 主動支持仿制藥開發和評估的一些戰略性、策略性措施值得重視,也對患者用藥的經濟可及產生了積極影響。其中包括:1)為生產商提供信息以開發和提交更高質量的申請,有助于提高競爭優勢;2)繼續實施FDA 的藥物競爭行動計劃(DCAP),提高仿制藥開發、審查和批準流程的效率;3)最大限度地提高復雜仿制藥的科學和法規透明度;4)為確保繼續獲得低成本、安全、有效和高質量的藥品做貢獻;5)加強國際合作,與30 多個監管機構共享信息,協調和促進科學和技術創新,從而最大限度地參與國際藥品監管計劃(IPRP)和ICH,進行監管交流,并確定潛在的仿制藥國際協調主題[57]。
為提高對仿制藥的國際戰略認識,筆者特別分析了近10 多年全球(美國數據)處方藥中仿制藥的情況(表6)。根據藥房報告的索賠數據,美國在2017 年編制了10 種最受歡迎的處方藥(Good Rx)清單,所有藥物都是通用的,這些處方有經濟可及、費用低的特點,大多數為仿制藥“老藥”,基本沒有相應年份批準的新藥。
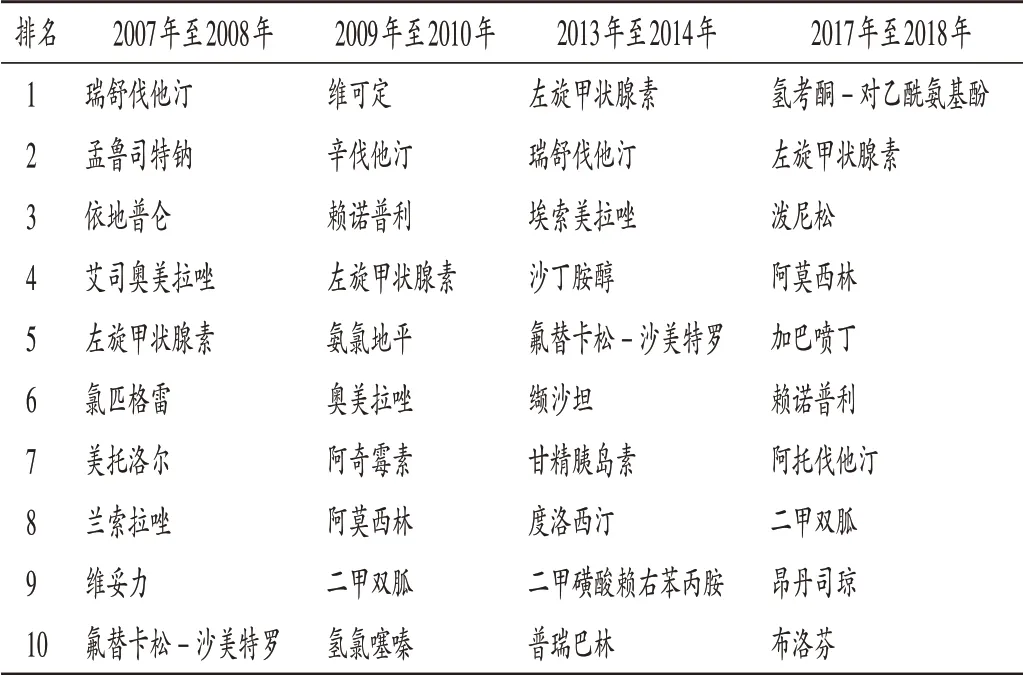
表6 美國不同年份處方量最大的10個藥物排名Tab.6 Ranking of the top 10 drugs with the largest number of prescriptions in the USA in different years
據統計,2021 年全球仿制藥市場規模超過4 000 億美元,其中美國是主戰場。專利藥專利到期是仿制藥市場增長的主要動力,預計未來仿制藥市場規模將以平均每年5%~10%的穩定速度增長,全球仿制藥行業依然處于較好的成長期。仿制藥發展增長的原因可能來自5 個方面:1)新藥研發效率低下,短期內難有突破;2)許多專利藥于2020 年至2025 年到期,讓仿制藥迎來戰略機遇期;3)世界經濟格局影響一些跨國大制藥企業轉向或涉足仿制藥開發,為此注入資金、擴大市場;4)世界各國醫療投入限制,降低醫療保障成本;5)全球生物仿制藥產業的發展動態,如歐美加速完善監管法規、美國平價醫療法案的落實,提供了較寬松的監管政策環境。
6 結語
從2020 年的銷售數據來看,隨著全球癌癥發病率的持續上升,2021 年抗腫瘤藥物繼續是發展的熱門領域。該類產品的特點為專利期長、靶點及仿制創新藥多且價格昂貴也會給企業研發帶來希望。從2021 年的實際發展情況來看,美國、歐盟和日本三大創新區塊所批準的新藥還是抗腫瘤藥物占絕對優勢。新藥研發是長周期、高投入、高風險的系統工程,對已有藥物的循證應用不可避免,已上市多年的現代藥物也紛紛出場,但COVID - 19 是一個全新的疾病領域,當前所有被應用于此病的藥物,自主增加新適應證的應用只能算是一種“同情用藥”。由于病毒的結構特點和變異特性,抗病毒藥物研發存在天然的困難,在科學和技術上還有許多未知問題,出現一種“神藥”并非易事。回望2021 年創新藥物的業績,關于COVID-19治療藥物的創新發展,可圈可點的并不多。
猜你喜歡
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
世界最新醫學信息文摘(2021年12期)2021-06-09 08:36:56
小學生優秀作文(低年級)(2018年6期)2018-05-19 01:54:28
消費導刊(2017年20期)2018-01-03 06:27:16
中國衛生(2016年6期)2016-11-23 01:09:08
中國衛生(2016年5期)2016-11-12 13:25:28
中國藥物應用與監測(2015年5期)2015-12-11 03:15:54
中國衛生(2015年9期)2015-11-10 03:11:14
中國衛生(2015年5期)2015-11-08 12:09:48
中國衛生(2015年4期)2015-11-08 11:15:58
