無序碳單層的電子結構及氫原子吸附的第一性原理研究
2022-03-04 02:32:38劉佳溪孫志海夏永鵬鄒勇進張煥芝黃鵬儒孫立賢
原子與分子物理學報 2022年4期
關鍵詞:結構
張 穎,劉佳溪,孫志海,黃 強,夏永鵬,李 彬,鄒勇進,張煥芝,黃鵬儒,徐 芬,孫立賢
( 桂林電子科技大學材料科學與工程學院 廣西信息材料重點實驗室,桂林 541004;廣西新能源材料構效關系協同創新中心,桂林 541004)
1 引 言
二維材料的探索與研究,能夠促進基礎科學的發展.二維材料因為其獨特的結構及優異的物理、化學特性,在多個領域具有潛在的技術應用價值.其中,石墨烯是一種蜂窩狀晶格排列的零帶隙半金屬二維材料,通過微機械剝離方法成功地從石墨中分離出來,這促使了研究人員對具有獨特性能的新型2D 材料的研究[1-3].然而,在石墨烯的生長或加工過程中,完美的蜂窩晶格往往會出現結構缺陷.例如Stone -Wales 缺陷[4],此晶體缺陷涉及到兩個π 鍵連接的碳原子的變化,即連接兩個碳原子的鍵圍繞鍵的中點旋轉90 度,可將四個六邊形轉換成兩個五邊形和兩個七邊形.
盡管缺陷的存在破壞了石墨烯晶格完美的對稱性,并影響了其某些性質,如降低載流子的遷移率[5,6],但缺陷對其他性質的影響是有益的.例如,缺陷可以增加石墨烯的反應活性,并可用于調整其在催化應用中的電子性能.因此,將非六邊形缺陷引入六邊形晶格是重建碳原子、改善電子性質的有效方法.目前,探索具有獨特晶體結構、機械和電子性能的新型二維碳的同素異形體的工作層出不窮[7-36].所提出的結構是由包含不同邊數的多邊形構成的,如4 + 8 環[7],5環[8],4 +6 +8 環[9,10,20-22],4 +5 +6 +8 +10環[11],5 +7 環[12-14],和5 +6 +7 環[13,15-19].然而,這些理論上提出的具有改進性質的二維碳的同素異形體多用于研究其力學性質和電學性質.因此,設計和探究新型的催化類二維碳的同素異形體具有重要的意義和價值.
表面吸附可以調節材料的電子結構,從而達到改變其表面性能的目的.特別是對于二維材料體系來說,它們擁有比其他材料較大的比表面積,可以通過吸附外來原子提高二維材料體系的化學活性.例如: 氫原子吸附于石墨烯可改變其電子和磁性結構.此外,氫原子與石墨化合物的相互作用在核聚變[37,38],氫儲存[39]等許多領域也有著重要作用.
在這項工作中,我們采用第一性原理計算方法研究了無序碳單層(5 +6 +8 +11 環) 的晶體結構、電子結構及其對氫原子吸附的性質,進而研究其催化性能.探討無序性導致的不同配位及局域結構對C 原子鍵長、原子電荷、電子分布的影響.通過在不同點位上進行氫原子吸附探討C 原子位點分布及化學活性.該研究將在一定程度上揭示二維無序碳材料結構-性能的構效關系,為實驗上設計新型催化類的無序碳功能材料提供理論指導.
2 模型構建與計算方法
基于密度泛函理論,本研究采用VASP 程序包進行計算[40-42].投影綴加波( PAW)[43,44]方法,用于處理電子-離子之間的相互作用,在改進的廣義梯度近似( GGA) 下,使用Perdew - Burke -Ernzerhof( PBE) 泛函形式來處理交換關聯函數.由于GGA 和局部密度近似LDA[45]不能準確的計算由于波動電荷分布之間的動力學相關性引起的范德華相互作用,本研究添加色散修正DFT -D3的方法來減小誤差[46,47].根據石墨烯固有缺陷的類型構建二維無序碳結構的初始結構.無序碳為全碳分子的二維平面網狀,晶格參數為a=b=17.22 ?,真空層c=10 ?.結構優化過程平面波的截斷能為500 eV,k-point 設置為6 ×6 ×1.體系的能量收斂標準為1 ×10-5eV.
為了研究了材料表面氫原子的吸附性能.無序碳吸附氫原子的吸附能定義如下:
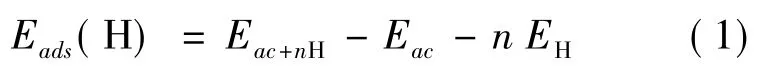
其中Eac+nH吸附H 原子后體系包含的總能量,Eac是無序碳的總能量,n 為吸附H 原子的數量,EH為自由的H 分子中H 原子的總能量.吸附能越負,對應的H 分子與H 原子吸附越強,模型越穩定.
對于給定模型的析氫性能( HER) 與單個氫原子的吸附能密切相關,同時吉布斯自由能越接近0,材料的析氫性能越優越.因此我們計算了吉布斯自由能,公式如下:
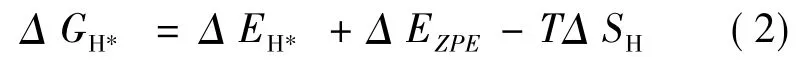
其中Δ EH*是吸附氫原子的能量,Δ EZPE是零點吸附能的變化.對于Δ SH,一般取Δ SH≈-1/2( Δ SH2) ,其中Δ SH2是氣相氫分子在標準條件下的熵.因此公式(2) 可以重新寫為[48]:
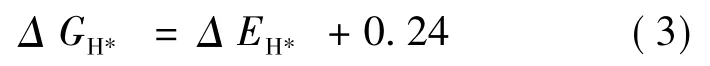
其中0.24 eV 是298 K 時ZPE 和熵的組合對表面模型的貢獻.通常認為吸附狀態下的氫吸附的振動熵可以忽略不計.
3 結果與討論
3.1 單層無序碳的幾何結構
圖1 為基于石墨烯的固有缺陷調節得到的無序碳結構.經優化體系達到穩定后,體系的形成能為34.475 eV,較大的形成能是因為體系內存在較多的斷鍵或較強的內部應力.石墨烯的碳碳鍵長為0.142 nm[48],但該體系中的碳碳鍵長與石墨烯中的鍵長有明顯不同,鍵長在0.13 -0.18 nm 之間,與石墨烯的碳碳鍵長相比最大的變化范圍達到26.7%.如圖1 所示,C-C 鍵被拉長的有C6- C20,C7- C22,C15- C29,C23- C35,C36-C48,C46- C47,C40- C41,C53- C68,C55- C69,C67-C80,C69-C84; 被壓縮的有C17-C18,C40-C41.因此,多元環的引入會導致附近碳原子的鍵長發生不同程度的拉伸或壓縮.原子間鍵長的變化會引起電化學性能的改變[48].為了研究無序多元環的引入對體系電子結構的影響,進一步計算了體系的電子態密度( DOS) 以及電子局域密度函數( ELF).
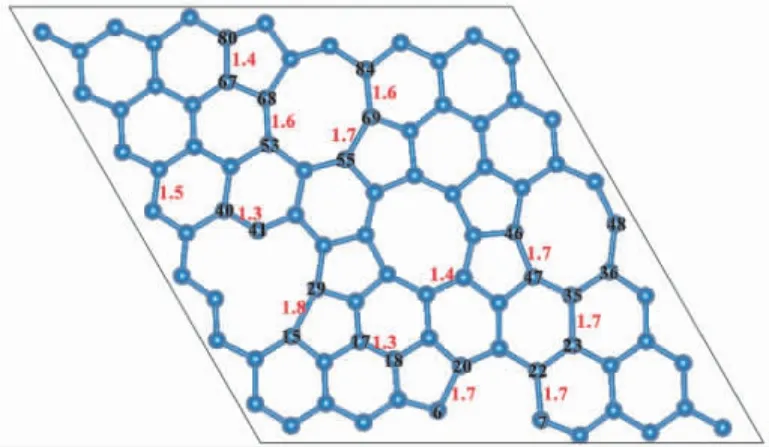
圖1 無序碳單層晶體結構Fig.1 Crystal structure of amorphous carbon monolayer
3.2 單層無序碳電子結構
在完美的六角晶格中,石墨烯在費米能級處形成狄拉克錐電子結構,電子有效質量小,遷移率大,使其呈現出類金屬的性質[49].六角晶格的破壞改變了這一電子結構,如圖2( a) 所示,在費米能級附近的錐形結構消失,出現較多局域電子峰,體系的電子態增加.這些電子峰的出現使得部分C 原子的電子局域性增強,化學活性發生改變.有研究報道,析氫活性起源于電子結構的變化,結構內電子態的改變可以為析氫反應提供豐富的活性位點[50].為此,本研究計算了單層無序碳的電子局域函數,考察不同位點處C 原子附近電子的局域特性及化學鍵性質的改變,如圖2( b)所示,可以看到,體系中電子主要集中在兩個C原子之間,例如C17- C18,C40- C41原子鍵長較短,約0.13 nm,其碳原子間的ELF 值≈0.95 表現出極強的共價鍵性質; 部分碳碳鍵長被拉伸處于0.16 ~0.18 nm 區間內( 如: C6- C20,C7-C22,C15- C29,C23- C35,C36- C48,C46- C47,C53-C68,C55-C69,C67-C80,C69-C84) ,其碳原子間的ELF 值相約為0.8 ~0.85.同時,在出現斷鍵又無法形成C -C 鍵的原子處會出現孤對電子,例如: 圖中C28原子只形成2 個配位,懸掛鍵為未配對電子,具有極強的局域性.C28的局域結構極不穩定,很容易通過吸附外來原子進行電子配對從而降低形成能.
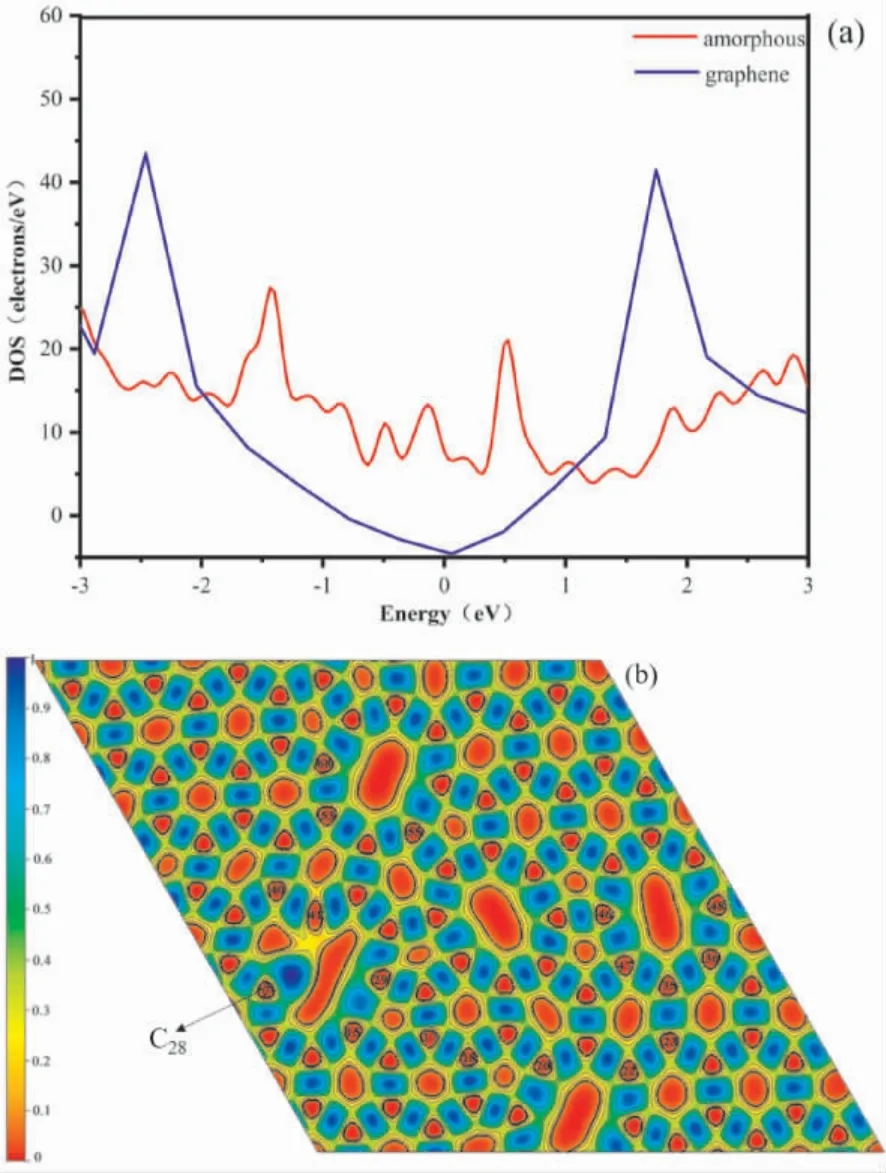
圖2 ( a) 無序碳單層與石墨烯的電子密度對比和( b) 電子局域密度函數Fig.2 ( a) Comparison of electronic density of states between amorphous carbonmonolayer and graphene; ( b) electronic local density function
圖3( a) 為單層無序碳結構的二維電荷密度分布情況.與圖2( b) 相比可以看出電荷密度分布與原子鍵長、電子局域性相對應.電荷密度相對越高,電子局域密度越大.體系中電荷聚集在C15-C16,C17-C18,C18- C19,C29- C30,C40- C41鍵長較短處,鍵長較長的碳碳鍵( C6- C20,C7-C22,C36-C48,C46-C47,C55-C69,C69-C84) 幾乎沒有電荷的聚集.不同鍵長的碳原子改變了體系的電子結構,增加了體系的聚集電荷和局域電子,使得不同碳原子位點可能表現出不同的電荷強弱.在電荷密度的基礎上對每個碳原子的Bader電荷進行了計算,Bader 電荷布居[51]是根據電荷零通量面分割的,通常認為該方法能夠較好的定義分配到原子上的電子數[52-54].圖3( b) Bader 電荷可以看出無序碳中的電子較多的原子為C20,C41,C67,失電子較多的原子的為C18,C21,C32.因為幾何結構變化的多樣性,結構內電子轉移呈現多樣性.圖3( c) 和( d) 分別是費米面下方和費米面上方區間的電荷分布圖.從圖中我們可以看出,最高占據態主要由C40,C41,C15,C14,C21,C22,C9等原子貢獻,最低未占據態主要由C15,C32,C21,C34,C52,C53等原子貢獻.從圖3( c)和( d) 中可看出,提供電子占據態的原子均位于多元環與六元環的交界位點.綜上所述,由于體系中局部六角晶格被破壞,無序性增加,改變了體系電子結構造成部分原子局域性增強.
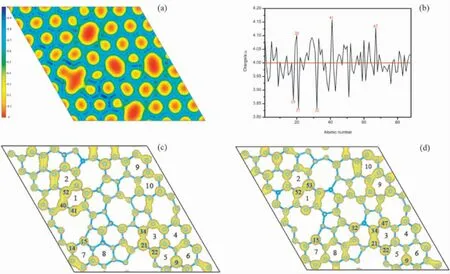
圖3 單層無序碳( a) 電荷分布情況; ( b) C 原子電荷布居; ( c) 費米面下方附近[-0.05,0]eV 區間的電荷密度; ( d) 費米面上方[0,0.05]eV 區間的電荷密度Fig.3 Amorphous carbon monolayer: ( a) charge distribution; ( b) C atom charge distribution; ( c) charge density in the[-0.05,0]eV interval near the bottom of the Fermi surface; ( d) above the Fermi surface[0 ,0.05]charge density in the eV interval
3.3 單層無序碳的氫吸附性能
上文中系統地分析了無序碳結構中的晶體結構和電子性質,發現C6,C20,C69等原子位點的碳碳鍵長較短,電荷聚集,電子局域,通常認為這些位點的化學活性較高.C15,C20,C40,C53,C68等原子位點碳碳鍵長較長,電子局域性較差,通常認為化學活性較低.接下來對這些位點的吸附性能進行定量的計算和研究.通過在相應的碳原子上方添加一個氫原子進行結構優化,如圖4所示為優化后得到的氫原子吸附結構圖,可以看出對于大部分的吸附點,氫原子吸附于碳原子的上方屬于頂位吸附,這主要是氫原子與碳原子的pz軌 道 成 鍵.例 如: C17,C20,C27,C32,C53,C68,C69.同時,一些位點的C -H 鍵會與體系處于同一平面,主要是因為氫原子可能與碳原子的sp2雜化軌道成鍵.可以看出吸附后C -H 鍵長與標準碳氫鍵( 0.109 nm)[55]相比變化范圍在0.0001 nm ~0.0028 nm 之間.吸附后氫原子位于無序碳平面內結構的C -H 鍵長較垂直于無序碳平面的C-H 鍵長變化較小,進一步說明氫原子占據sp2軌道形成的C-H 鍵鍵強較強.
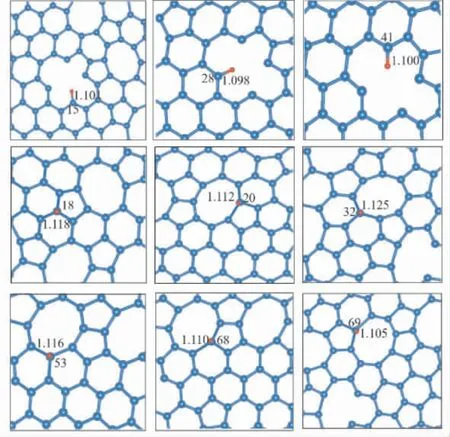
圖4 單層無序碳氫原子吸附的俯視圖模型Fig.4 Hydrogen adsorption model of amorphous carbon monolayer
圖5 為吸附前后態密度對比圖.不同位點氫原子吸附后總態密度峰的位置發生變化,在費米能級附近H( s) 與C( p) 軌道發生大部分的重合與少量的雜化.吸附后體系的總態密度較吸附前產生較大的差異,以C15為例費米能級附近的局域電子峰減少.C15,C28兩位點氫原子的態密度曲線均有一個較高的峰值,氫原子軌道占據較高; 其他位點如C17,C20等吸附后氫原子的態密度較平坦,離域較大.氫原子的成鍵方式以及軌道的占據情況會對其吸附能造成一定的影響,如圖6( a)C15,C28兩個位點的吸附能分別為-1.76 eV, -2.21 eV 較其他吸附位點吸附能低,吸附后結構更穩定.單個原子的吸附能與解離能的強弱是析氫反應發生的關鍵[56-60]圖5、圖6( a) 以及表1的數據綜合分析,不同位點不同鍵長以及鍵角對氫原子的吸附能力不同.例: C15,C28,C40三個位點呈現出二配位結構,對氫原子的吸附能較大,但其過大的吸附能導致氫原子的解離相對較難;C68,C69二者在多元環結構中所處位置的鍵角較大,導致其吸附能較大; C17,C18兩原子位于五元環與六圓環之間且鍵角較平均,對氫原子的吸附能力較弱.綜上,通過調節共價鍵的性質可以調整其成鍵方式,不同的成鍵方式會影響其吸附氫原子的能量.氫原子的成鍵方式不同,導致各位點的吸附能以及解離能不同,從而為析氫反應產生更多的活性位點.
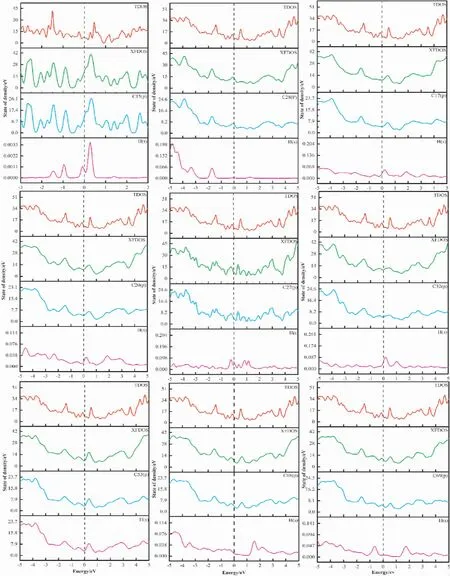
圖5 吸附前的總態密度與各位點吸附后總態密度、各吸附碳原子以及氫原子態密度的對比圖Fig.5 Comparison diagrams of the total density of states before adsorption and that after adsorption on each site,the densities of states of each adsorbed carbon atom and hydrogen atom.
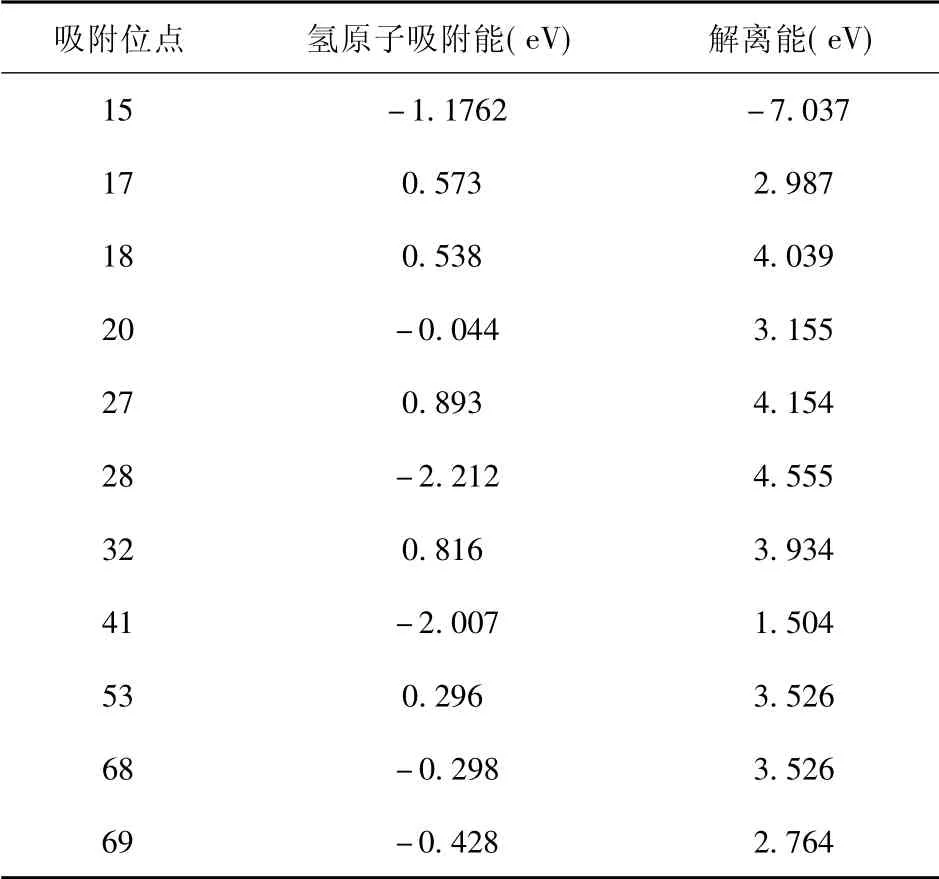
表1 不同位點氫原子解離能Table 1 Dissociation energies of hydrogen atom at differentsites
為了更好地探索無序碳材料析氫反應的性能,計算了不同位點對氫吸附的吉布斯自由能如圖6( b).對于良好的析氫反應催化劑,如Pt 的ΔGH*值接近于零,其代表著對于氫原子的吸附和釋放強度適中[58].圖6( b) 中C53,C68位點的吉布斯自由能與Pt 的吉布斯自由能接近.兩位點均為三配位具有較大的鍵角且pz軌道與氫原子成鍵.因此無序碳材料可以通過自身電子結構變化引起活性位點的增加為析氫反應提供良好的催化劑.
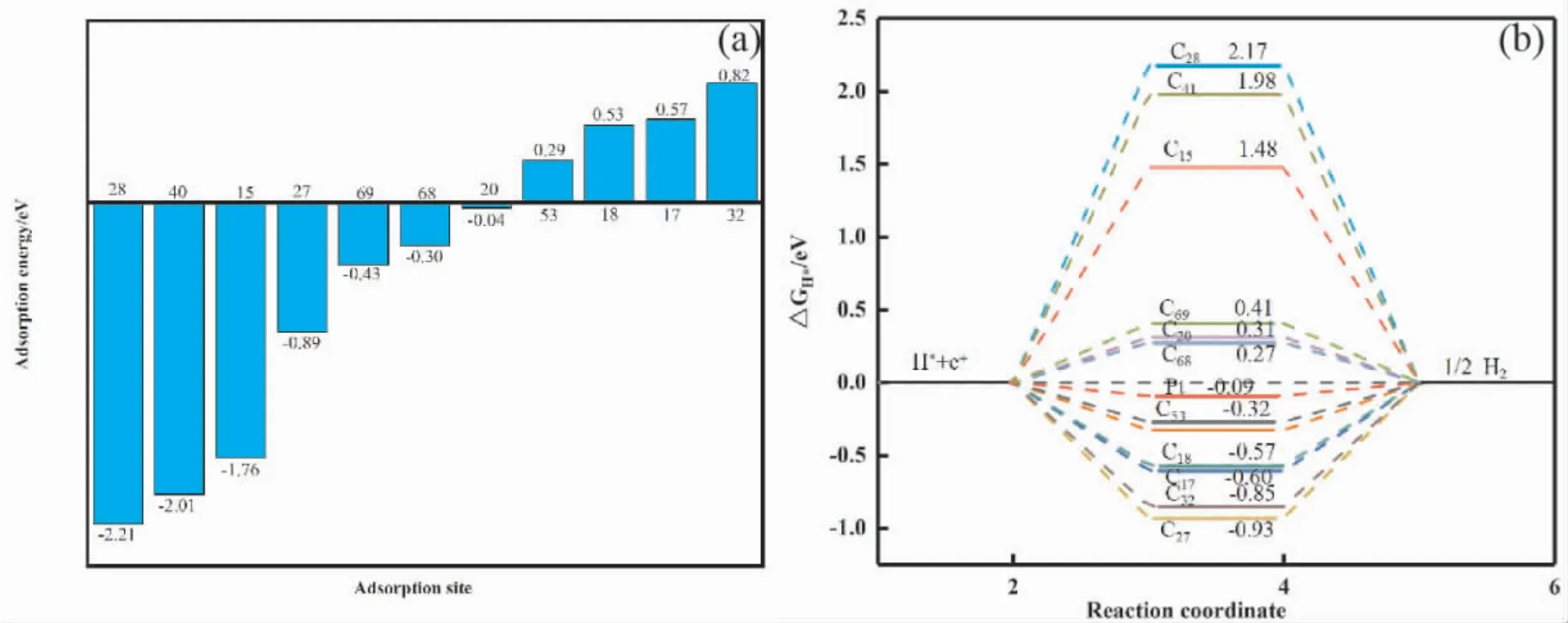
圖6 無序碳單層( a) 不同吸附位點的吸附能和( b) 無序碳吸附氫原子的吉布斯自由能Fig.6 Amorphous carbon monolayer ( a) adsorption energies of different adsorption sites and ( b) Gibbs free energies
4 結 論
本研究運用基于密度泛函理論第一性原理計算研究了二維單層無序材料的幾何結構、電子結構及其析氫性能,得到如下結論:
1) 體系中缺陷的引入,改變了部分位點的鍵長、電荷密度以及電荷轉移量和電子局域性,使材料中增加了大量的性能各異的吸附位點,例如:C53,C68,C15,C28等.
2) 通過對不同位點晶體結構和吸附性能的對比,發現C53,C68吸附氫適中具有較好催化潛力.這些位點的特征是與相鄰碳原子具有三個配位,并且三個sp2共價鍵的對稱性被打破,其中一個鍵角增大.
3) 通過調控在六元環與多元環交界處構造三配位且鍵角較大的位點可以調節吸附氫性能使其具有較好的析氫性能.
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
