低硅X分子篩的合成研究
2022-02-15 11:42:24高寧寧劉相李拓鵬飛高俊魁王輝國
石油煉制與化工 2022年2期
關鍵詞:體系
高寧寧,劉相李,拓鵬飛,高俊魁,鐘 進,王輝國,郁 灼
(中國石化石油化工科學研究院,北京 100083)
工業上X分子篩可作為對二甲苯吸附劑的活性組分,用于從混合C8芳烴中吸附分離高純度對二甲苯產品。研究表明,X分子篩選擇性吸附對二甲苯的性能可能源于其超籠中的特定靜電場[1-2],硅鋁比[n(SiO2)/n(Al2O3)]是影響超籠內靜電場性質的關鍵因素,因而X分子篩選擇性吸附對二甲苯的性能與其硅鋁比密切相關。王輝國等[3]采用脈沖試驗研究了BaX分子篩的硅鋁比對C8芳烴分離系數的影響,發現隨著分子篩硅鋁比降低,對二甲苯相對于間二甲苯和鄰二甲苯的分離系數先升高后降低,而對二甲苯相對于乙苯的分離系數逐漸增加。因此,通過降低X分子篩的硅鋁比來調變其分離性能,尤其是提高對二甲苯相對于乙苯的吸附選擇性成為重要的研究方向。
X分子篩的拓撲結構為FAU,硅鋁比為2.0~3.0[4]。通常情況下,X分子篩合成體系以氫氧化鈉為堿源,但當合成體系的硅鋁比較低時,晶化產物易形成A分子篩雜晶[5];若以氫氧化鈉和氫氧化鉀為混合堿源,則可以在更寬范圍內調變合成體系的硅鋁比,所得X分子篩的硅鋁比可低至2.0[6]。Iwama等[7]認為,在水熱條件下合成體系液相中存在大量硅鋁比為2.0的四元環結構;在鉀離子作用下,這些四元環結構組裝形成X分子篩的次級結構單元雙六元環,抑制了A分子篩成核。趙東璞等[8]同樣發現在合成體系中引入鉀離子可有效抑制A分子篩雜晶形成。因此,合成體系中的鉀離子對低硅X分子篩骨架結構的形成有重要影響。
低硅X分子篩可以采用低溫老化和高溫晶化的兩段法合成[9-10]。低溫老化階段,合成體系中的硅物種、鋁物種發生可逆縮合反應,在Si—O—Si、Si—O—Al不斷形成與斷裂的動態過程中逐漸形成晶核[11],老化溫度對晶核的種類和數量產生影響,進而影響分子篩純度;高溫晶化階段,硅物種、鋁物種在晶核表面發生縮合-再溶解反應,晶核逐漸長大形成三維骨架結構,晶核表面縮合反應的種類和速率受晶化溫度影響,晶化溫度不適宜也會導致產物中形成雜晶[12-13]。
為了獲得結晶度高、雜晶少的低硅X分子篩,本研究以氫氧化鈉和氫氧化鉀的混合物作為合成體系堿源,系統研究合成體系硅鋁比、堿硅比[n(Na2O+K2O)/n(SiO2) ]、水堿比[n(H2O)/n(Na2O+K2O)]、鉀堿比[n(K2O)/n(Na2O+K2O)]以及老化溫度、晶化溫度對產物結晶度、甲苯吸附容量等的影響,并采用X射線衍射(XRD)、X射線熒光光譜(XRF)、掃描電鏡(SEM)、29Si魔角旋轉固體核磁共振(29Si MAS NMR)、27Al魔角旋轉固體核磁共振(27Al MAS NMR)等手段表征產物的理化性質。
1 實 驗
1.1 原 料
氫氧化鈉,分析純,天津市大茂化學試劑廠產品;氫氧化鉀,優級純,阿拉丁試劑公司產品;水玻璃,中國石化催化劑有限公司齊魯分公司產品;低堿度偏鋁酸鈉,自制。
1.2 低硅X分子篩合成
按照體系物料比:n(SiO2)/n(Al2O3)=2.05、n(Na2O+K2O)/n(SiO2)=3.25、n(H2O)/n(Na2O+K2O)=17、n(K2O)/n(Na2O+K2O)=0.23,將氫氧化鈉和氫氧化鉀溶解在去離子水中,在強烈攪拌下依次加入水玻璃、低堿度偏鋁酸鈉,繼續攪拌30 min,得到乳白色均勻合成體系;然后,將其轉移至密閉水熱反應釜中,在靜態條件下于70 ℃老化6 h,之后于95 ℃晶化3 h;晶化產物經過濾、洗滌至pH為8~9,在120 ℃干燥后得到低硅X分子篩。
為考察單一因素對產物性質的影響,在上述物料配比、溫度、時間條件下,保持其他因素不變,分別調整硅鋁比在1.90~2.65、堿硅比在2.25~5.00、水堿比在10~32、鉀堿比在0~0.53、老化溫度在35~80 ℃、晶化溫度在70~150 ℃間變化,得到的分子篩產物分別記作A-a,B-b,C-c,D-d,E-e,F-f,其中a,b,c,d,e,f分別為其他因素不變條件下合成體系中硅鋁比、堿硅比、水堿比、鉀堿比、老化溫度、晶化溫度的數值。
1.3 表 征
采用PHILIPS公司生產的X’Pert型X射線衍射儀測定分子篩樣品的X射線衍射譜圖,Cu靶,管電壓為40 kV,管電流為40 mA,2θ掃描范圍為5°~50°,掃描步長為0.02°。分子篩的相對結晶度(R)為測試樣品XRD圖譜中2θ為10.0°±0.1°,11.7°±0.1°,15.4°±0.1°,23.3°±0.1°,26.6°±0.1°,30.9°±0.1°處衍射峰高度之和與參比樣品(A-2.05)在對應位置衍射峰高度之和的比值[14]。
分子篩樣品的元素分析采用日本理學株式會社生產的ZSX Primus ⅡX射線熒光光譜儀測定,電壓為50 kV,電流為50 mA。分子篩樣品的形貌采用Hitachi公司生產的S-4800型掃描電子顯微鏡表征,加速電壓為5 kV,電流為10 μA。
分子篩樣品的29Si MAS NMR圖譜在Bruker AVANCE Ⅲ 500 MHz核磁共振儀上測定,采用7 mm的ZrO2轉子,轉速為5 kHz,脈沖寬度為2.03 μs,采樣次數為3 000。27Al MAS NMR圖譜在Bruker AVANCE Ⅲ 600 MHz共振儀上測定,采用4 mm的ZrO2轉子,轉速為12 kHz,脈沖寬度為0.51 μs,采樣次數為5 000。
分子篩樣品的甲苯吸附容量在自建裝置上用動態法測定。首先將樣品于500 ℃焙燒2 h,再將焙燒后的樣品置于干燥器中降至室溫,然后稱量樣品質量(m1),在35 ℃下,用氮氣將甲苯蒸氣攜帶入樣品室,甲苯蒸氣相對壓力(p/p0)為0.5,16 h后稱量吸附甲苯后的樣品質量(m2)。待測分子篩樣品的甲苯吸附容量(W)為(m2-m1)/m1。
2 結果與討論
2.1 合成體系物料配比的優化
2.1.1硅鋁比
其他條件不變,硅鋁比分別為1.90,2.05,2.20,2.35,2.50,2.65時所得產物的XRD圖譜如圖1所示。
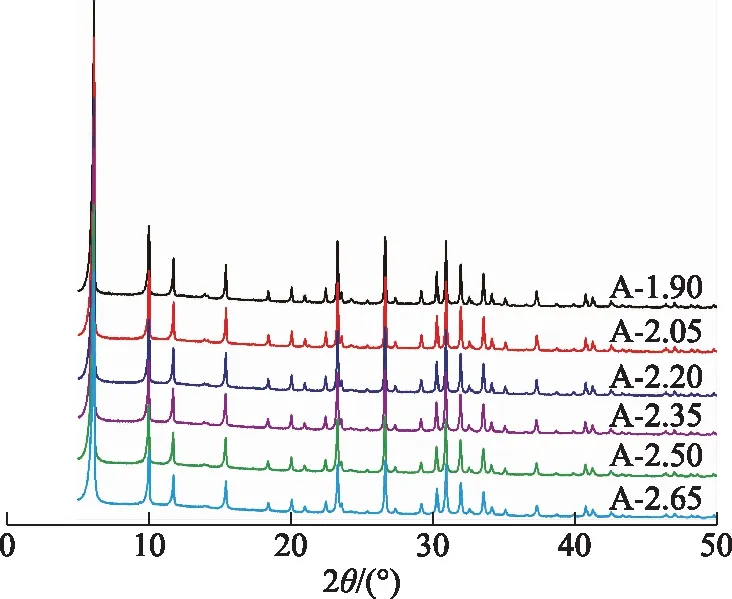
圖1 合成體系中不同硅鋁比下所得產物的XRD圖譜
由圖1可以看出:A-1.90在2θ為6.1°,10.0°,11.7°,15.4°,23.3°,26.7°,31.0°附近出現較強的X分子篩特征衍射峰,表明產物中生成了大量的X分子篩[15-16];此外在2θ為7.2°附近出現微弱的衍射峰,表明產物中有極少量A分子篩雜晶形成[17]。通常情況下,A分子篩合成體系的硅鋁比小于或等于2.0,晶化溫度為90 ℃左右[18]。因此,當低硅X分子篩合成體系的硅鋁比低于2.0時,產物中容易形成A分子篩。當合成體系的硅鋁比從2.05逐漸升至2.65時,所得產物均為純的X分子篩,表明當合成體系的硅鋁比為2.05~2.65時均可以合成出純X分子篩。
合成X分子篩的相對結晶度及甲苯吸附容量隨合成體系硅鋁比變化的曲線如圖2所示。由圖2可以看出,合成體系硅鋁比從1.90增加到2.05時,X分子篩的結晶度從97%增至100%,合成產物的甲苯吸附容量從200 mg/g增至203 mg/g。依據Loewenstein規則[19],分子篩的骨架硅鋁比最低為2.00,當合成體系硅鋁比低于2.00時,過量的鋁物種難以進入分子篩骨架結構,可能以無定形鋁的形式存在于產物中,且產物中存在少量A分子篩雜晶,從而導致A-1.90的結晶度和甲苯吸附容量均稍低于A-2.05。當合成體系的硅鋁比繼續增加至2.65時,X分子篩的結晶度和甲苯吸附容量均逐漸下降。這可能是因為,當合成體系的硅鋁比大于2.05、且合成體系的堿硅比保持3.25時,較高的堿濃度不利于X分子篩晶化,導致X分子篩的結晶度和合成產物的甲苯吸附容量下降。因此,合成體系的最佳硅鋁比為2.05。
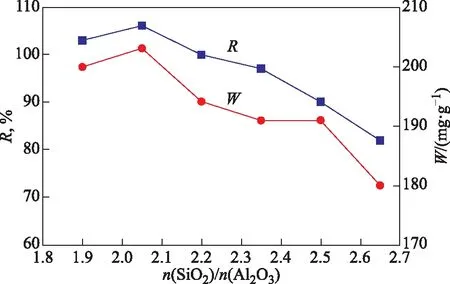
圖2 合成X分子篩的結晶度和甲苯吸附容量隨合成體系硅鋁比的變化曲線
2.1.2堿硅比
其他條件不變,合成體系的堿硅比分別為2.25,2.75,3.25,4.00,4.50,5.00時所得產物的XRD圖譜如圖3所示。由圖3可以看出,當合成體系堿硅比為2.25~5.00時,所得產物均為純X分子篩,說明在合成體系其他條件不變,堿硅比在較寬范圍內變化均能夠獲得純X分子篩。
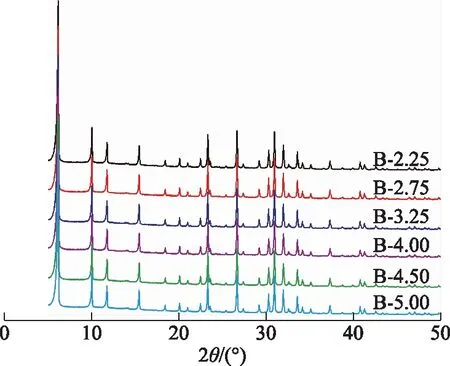
圖3 合成體系中不同堿硅比下所得產物的XRD圖譜
合成X分子篩的相對結晶度和甲苯吸附容量隨合成體系堿硅比的變化曲線見4。由圖4可以看出,隨著合成體系堿硅比從2.25增至5.00,合成的系列X分子篩樣品的結晶度和甲苯吸附量呈現出相似的變化趨勢:結晶度先從86%升至100%再降至92%,甲苯吸附容量先從194 mg/g升至203 mg/g再降至197 mg/g。這可能是因為當合成體系的堿硅比低于3.25時,合成體系中的硅、鋁物種難以完全結晶;而當合成體系的堿硅比高于3.25時,較高的堿濃度會對分子篩的晶體結構造成破壞,進而導致產物的結晶度和甲苯吸附容量降低。這與王輝國等[20]的研究結果一致,合適的堿硅比有利于獲得結晶度較高的X分子篩。因此,合成體系優選的堿硅比為3.25。

圖4 合成X分子篩的結晶度和甲苯吸附容量隨合成體系堿硅比的變化曲線
2.1.3水堿比
其他條件不變,合成體系水堿比分別為10,12,17,22,27,32時所得產物的XRD圖譜如圖5所示。由圖5可以看出:C-10在2θ為13.9°和24.3°附近出現較強的衍射峰,表明產物中僅形成了SOD分子篩[21];C-12中含X分子篩和SOD分子篩,且SOD分子篩對應的衍射峰強度減弱;C-17為純的X分子篩;C-22中除了形成大量X分子篩,還出現少量A分子篩;C-27中也形成了大量X分子篩,但A分子篩的衍射峰強度增強;C-32中僅形成了X分子篩,但是其對應的衍射峰強度顯著減弱。這表明:當合成體系的水堿比小于17時,產物中容易形成SOD分子篩雜晶,且隨著水堿比的增大,產物中X分子篩增多,SOD分子篩雜晶減少;當合成體系的水堿比大于17時,產物中容易形成A分子篩雜晶,且隨著水堿比繼續增大,A分子篩雜晶增多;當合成體系的水堿比達到32時,僅形成了少量X分子篩,這可能是因為合成體系中堿濃度太低,導致硅、鋁物種的晶化難度增大。

圖5 合成體系中不同水堿比下所得產物的XRD圖譜
合成X分子篩的相對結晶度和甲苯吸附容量隨合成體系水堿比的變化曲線見圖6。由圖6可以看出,當合成體系的水堿比從10增至17時,X分子篩的結晶度從0升至100%,甲苯吸附容量從20 mg/g逐漸升至203 mg/g。這是因為隨著合成體系水堿比的增大,產物中X分子篩的含量逐漸增加,SOD分子篩雜晶的含量逐漸降低。當合成體系的水堿比繼續增加至32時,產物中A分子篩雜晶的含量增加;尤其是當水堿比為32時所得產物中X分子篩的含量非常低,導致X分子篩的結晶度和甲苯吸附容量快速下降。因此,合成體系優選的水堿比為17。
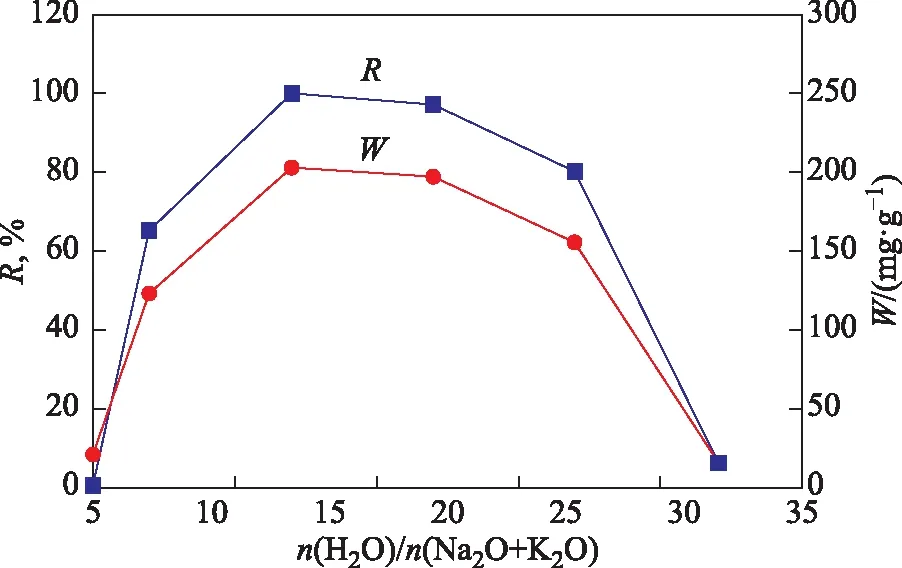
圖6 合成X分子篩的結晶度和甲苯吸附容量隨合成體系水堿比的變化曲線
2.1.4鉀堿比
其他條件不變,合成體系鉀堿比分別為0,0.13,0.23,0.33,0.43,0.53時所得產物的XRD圖譜如圖7所示。由圖7可知:D-0為SOD分子篩;D-0.13主要為X分子篩,并包含A分子篩和SOD分子篩,其中SOD分子篩衍射峰強度較D-0明顯降低,說明其含量較低;D-0.23,D-0.33,D-0.43均為純X分子篩;D-0.53也為純X分子篩,但其衍射峰強度大幅降低。這說明在合成體系中加入一定量的氫氧化鉀可以抑制A分子篩雜晶和SOD分子篩雜晶的形成,有利于獲得純X分子篩產物,但鉀含量過高對X分子篩的形成不利。這與Iwama等[6]的研究結果一致。
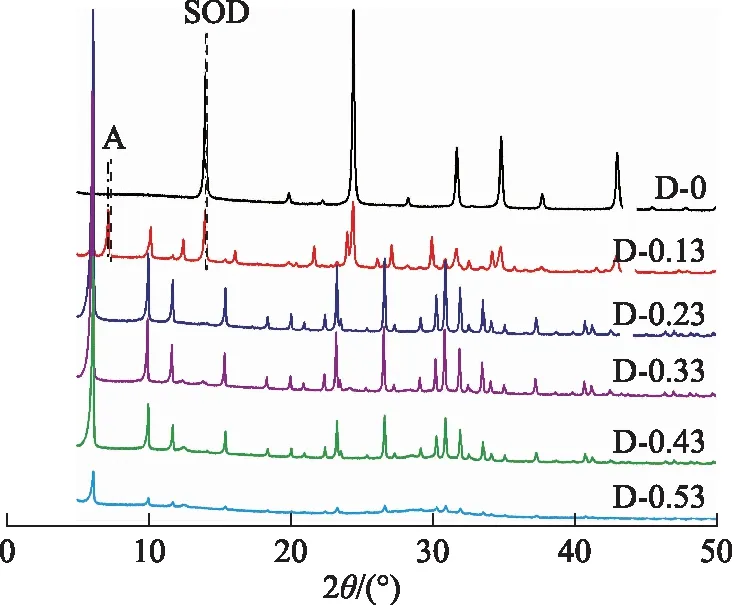
圖7 合成體系中不同鉀堿比下所得產物的XRD圖譜
合成X分子篩的相對結晶度和甲苯吸附容量隨合成體系鉀堿比的變化曲線見圖8。由圖8可以看出:鉀堿比為0時,所得產物為SOD分子篩,此時以X分子篩計的結晶度和合成產物的甲苯吸附容量均為0;鉀堿比增加至0.13時,X分子篩結晶度有所增加,但是產物中仍然包含明顯的A分子篩雜晶和SOD分子篩雜晶,導致其甲苯吸附容量仍然較低;當鉀堿比增至0.23時,X分子篩的結晶度和甲苯吸附容量均達到最高點,分別為100%和203 mg/g;繼續增加鉀堿比至0.53,X分子篩的結晶度和甲苯吸附容量均快速降低。這是因為合成體系中鋁物種濃度隨著鉀含量的升高而降低,鋁物種和鈉離子的濃度較低會降低成核和晶體生長速率[22],不利于產物中X分子篩結晶度的提高。因此,合成體系優選的鉀堿比為0.23。

圖8 合成X分子篩的結晶度和甲苯吸附容量隨合成體系鉀堿比的變化曲線
綜上所述,合成體系的硅鋁比、堿硅比、水堿比和鉀堿比對合成產物的物相組成、X分子篩的結晶度和合成產物的甲苯吸附容量有不同程度的影響。比較而言,保持合成體系的水堿比為17、鉀堿比為0.23不變,在較寬范圍內調變合成體系的硅鋁比和堿硅比時不易產生雜晶,但所得X分子篩的結晶度和甲苯吸附容量存在差異;而改變體系的水堿比或鉀堿比,產物中易形成SOD或A分子篩雜晶。當合成體系的水堿比小于或等于12時,產物中易形成SOD分子篩雜晶;水堿比超過22時,產物中易形成A分子篩雜晶;水堿比過高(達32)時,則會導致分子篩結晶困難。當合成體系的鉀堿比大于或等于0.23時,可以有效抑制SOD和A分子篩雜晶的形成,獲得純X分子篩;但鉀堿比過高時,則不利于X分子篩晶化。因此,合成體系優選的硅鋁比為2.05、堿硅比為3.25、水堿比為17、鉀堿比為0.23。
2.2 低硅X分子篩合成溫度
2.2.1老化溫度
其他條件不變,分別在35,50,60,70,80 ℃老化、再在95 ℃晶化后所得產物的XRD圖譜,以及不經老化而直接在95 ℃晶化后所得產物(E-95)的XRD圖譜如圖9所示。由圖9可以看出,E-35包含X分子篩和少量A分子篩雜晶,E-50,E-60,E-70,E-80均為純X分子篩,E-95包含X分子篩和少量SOD分子篩雜晶。表明老化溫度為50~80 ℃時均能夠合成出純X分子篩;老化溫度較低時(低于50 ℃)容易形成A分子篩雜晶;不經過老化而直接在95 ℃晶化,容易形成SOD分子篩雜晶。X,A,SOD分子篩骨架結構中均包含α籠結構單元,區別在于其相鄰α籠之間的連接方式不同:X分子篩中的相鄰α籠由雙六元環相連,A分子篩中的相鄰α籠由雙四元環相連,SOD分子篩中的相鄰α籠直接通過四元環相連。因此,推斷老化溫度低于50 ℃時易形成雙四元環,老化溫度為50~80 ℃時促進雙六元環的形成;不經老化而直接95 ℃晶化處理,不利于形成雙六元環和雙四元環,而是導致形成更為致密的SOD分子篩。
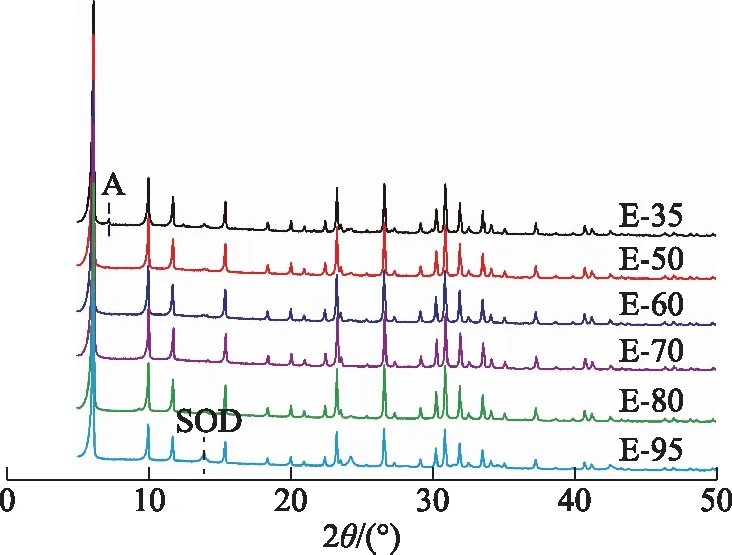
圖9 不同老化溫度下所得產物的XRD圖譜
合成產物X分子篩的相對結晶度和甲苯吸附容量隨老化溫度的變化曲線見圖10。由圖10可以看出:老化溫度為35 ℃時,X分子篩的結晶度和合成產物的甲苯吸附容量較低,分別為95%和188 mg/g,原因在于產物中存在少量A分子篩雜晶;老化溫度達到50~80 ℃時,X分子篩的結晶度和合成產物的甲苯吸附容量較高,分別為99%~100%和202~203 mg/g;不經過老化而在95 ℃直接晶化,使X分子篩的結晶度和合成產物的甲苯吸附容量快速降低,這是因為所得產物中出現SOD分子篩雜晶。老化階段是分子篩晶核的主要形成時期,因而老化處理可以有效提高晶化的速率,從而提高產物中X分子篩含量[5,8]。
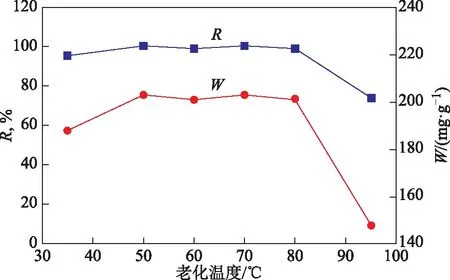
圖10 合成X分子篩的結晶度和甲苯吸附容量隨老化溫度的變化曲線
2.2.2晶化溫度
其他條件不變,晶化溫度分別為70,95,115,130,150 ℃時所得產物的XRD譜圖如圖11所示。由圖11可以看出:F-70和F-95均僅含X分子篩,表明晶化溫度為70~95 ℃時均能夠合成出純X分子篩;F-115中除了形成大量X分子之外篩還出現少量SOD分子篩雜晶,F-130中的SOD分子篩含量有所增加,表明晶化溫度為115 ℃時,產物中開始出現SOD分子篩,且SOD分子篩含量隨著晶化溫度的升高而增加;晶化溫度達到150 ℃時,產物為純SOD分子篩,說明晶化溫度過高時不能形成X分子篩。這可能是因為SOD分子篩較X分子篩更為致密,而在較高晶化溫度下易形成致密相[23]。
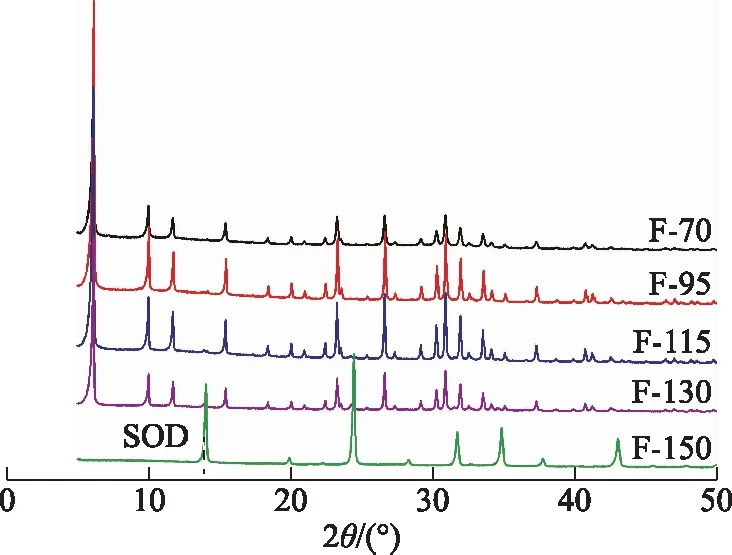
圖11 不同晶化溫度下所得產物的XRD圖譜
合成產物X分子篩的相對結晶度和甲苯吸附容量隨晶化溫度的變化曲線見圖12。由圖12可以看出:隨著晶化溫度從70 ℃升至150 ℃,X分子篩的結晶度和合成產物的甲苯吸附容量均呈現先升高后降低趨勢;晶化溫度為95 ℃時,X分子篩結晶度和甲苯吸附容量最高。表明雖然晶化溫度在70~95 ℃時均能夠合成出純X分子篩,但產物質量存在差異。溫度過低(如70 ℃)時,可能由于晶化速率較小而導致合成產物不能完全晶化;而溫度超過115 ℃時,產物中SOD分子篩雜晶含量逐漸增加,導致合成產物的甲苯吸附容量降低。因此,較優的晶化溫度為95 ℃。
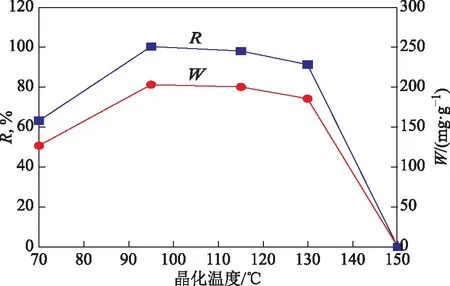
圖12 合成X分子篩的結晶度和甲苯吸附容量隨晶化溫度的變化曲線
綜上所述,在較寬的老化和晶化溫度范圍內,均可以合成出純X分子篩;晶化之前進行老化處理是必要的,不經老化處理而直接晶化會導致產物中出現SOD分子篩雜晶,老化溫度較低時容易引入A分子篩雜晶;晶化溫度較低時會降低X分子篩的晶化速率,而晶化溫度過高則容易形成SOD分子篩雜晶。
2.3 低硅X分子篩的元素與形貌表征
基于以上研究結果,在優化的條件下合成了低硅X分子篩,并采用XRF、核磁共振波譜、SEM等手段進行表征。由XRF表征結果可知,所得低硅X分子篩的硅鋁比為1.96,陽離子為鉀和鈉,鉀堿比為0.24,與合成體系的鉀堿比接近。
低硅X分子篩的29Si MAS NMR和27Al MAS NMR表征結果如圖13所示。由圖13(a)可知,低硅X分子篩在化學位移為-84附近出現強峰,在化學位移為-89附近有非常弱的寬峰,分別對應于分子篩骨架結構中的Si(4Al)和Si(3Al)。表明低硅X分子篩骨架結構基本由Si(4Al)組成,僅含極少量的Si(3Al),經擬合計算可知其骨架硅鋁比為2.00[24],與XRF的表征結果一致。由圖13(b)可知,低硅X分子篩在化學位移63附近出現強峰,表明樣品中的鋁均以4配位形式存在,不含非骨架鋁[14]。
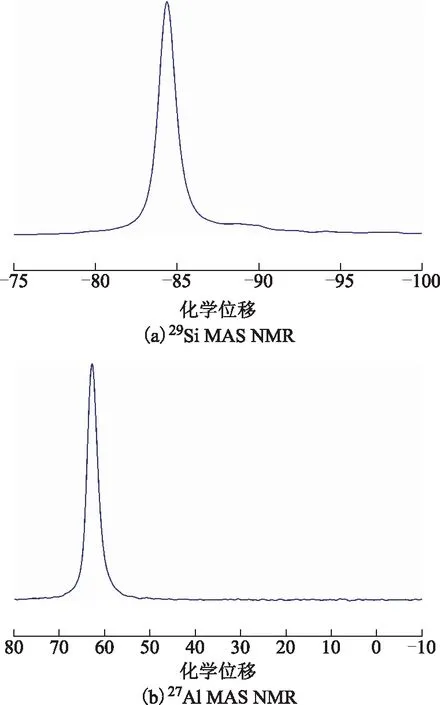
圖13 低硅X分子篩的29Si MAS NMR和27Al MAS NMR圖譜
圖14為低硅X分子篩的SEM照片。由圖14可以看出,低硅X分子篩為直徑2~5 μm球體,表面凹凸不平,含大量尺寸為0.5~1.5 μm的四面體突起。由于X分子篩通常呈八面體形貌[11],因此可知球體結構是由大量尺寸為0.5~1.5 μm的X分子篩晶粒聚集而成。

圖14 低硅X分子篩的SEM照片
3 結 論
合成低硅X分子篩體系優化的硅鋁比、堿硅比、水堿比、鉀堿比分別為2.05,3.25,17,0.23,優化的老化溫度為50~80 ℃、晶化溫度為95 ℃。在優化條件下,可合成出結晶度較高的純低硅X分子篩。合成的低硅X分子篩不含非骨架鋁,硅鋁比為2.0,外觀呈直徑2~5 μm的球體形貌,球體由尺寸為0.5~1.5 μm的X分子篩晶粒聚集而成。
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
