HPLC 雙波長法同時測定桑白皮水提物中6 個成分的含量*
2022-02-02 13:08:14戴麗娜陳泳君
藥學(xué)與臨床研究 2022年6期
戴麗娜,陳泳君,楊 穎
連云港市食品藥品檢驗檢測中心,連云港222006
桑白皮為桑科植物桑(Morus alba L.)的干燥根皮,又名桑皮、桑根白皮,始記載于《神農(nóng)本草經(jīng)》,具有瀉肺平喘及利水消腫的功效[1],是臨床常用的中藥材之一。近年來,有研究表明,桑白皮提取物具有美白功效,且因其毒性很低,相比于化學(xué)美白劑具有更高的安全性,同時桑樹在我國廣泛栽種,具有豐富的應(yīng)用資源,成本低廉,從而在化妝品美白領(lǐng)域得到越來越多的關(guān)注,具有廣闊的研發(fā)前景[2,3]。對于桑白皮中的有效美白成分,需要建立合適的提取方法,以達(dá)到既能有效地提取最大含量的活性組分,又不危害人員的身體健康、不污染環(huán)境的目的。通過查閱桑白皮的研究文獻[4,5],主要通過水、甲醇和乙醇進行不同程度的萃取。甲醇屬于《化妝品安全技術(shù)規(guī)范(2015 年版)》中的禁用物質(zhì),即不得添加使用,但其可能作為其它化妝品原料的雜質(zhì)被帶入,故該規(guī)范規(guī)定化妝品中甲醇含量不得超過2000 mg·kg-1。乙醇作為一種有機溶劑,它會溶解皮脂膜甚至細(xì)胞間脂質(zhì)中的油脂,造成皮膚屏障功能被破壞,對于含有乙醇的護膚品,一向存在爭議。綜合上述情況,本研究選取水作為提取溶劑。研究發(fā)現(xiàn),桑白皮化學(xué)成分主要為黃酮、苷以及其他類型化合物[6]。2020 年版《中國藥典》未對桑白皮成分含量測定項作出規(guī)定。本研究建立同時測定桑白皮水提物中6個主要成分(桑皮苷A、綠原酸、單寧酸、桑根酮D、桑酮G、桑根酮C)含量的方法,以期為桑白皮水提物中的成分進行測定提供依據(jù)。
1 儀器與藥品、試劑
1.1 儀器
Thermo U3000 高效液相色譜儀,二極管陣列檢測器(賽默飛世爾科技有限公司);BT125D 電子分析天平(德國賽多利斯公司);KQ-300DE 型超聲波清洗器(昆山超聲波儀器有限公司)。
1.2 藥品與試劑
對照品:綠原酸(批號:110753-202018,純度:96.1%,購自中國食品藥品檢定研究院);單寧酸(批號:040106-202203,純度:98.46%,上海鴻永生物科技有限公司);桑皮苷A(批號:JOT-11177,純度:99.22%)、桑根酮C(批號:JOT-11145,純度:90.90%)、桑根酮D(批號:JOT-11146,純度:94.62%)、桑酮G(批號:JOT-11265,純度:99.11%,自成都普菲德生物技術(shù)有限公司)。
乙腈(色譜級,德國Merck 公司);甲酸為色譜級;水為超純水。
桑白皮藥材(來源:江蘇仟草堂藥業(yè)有限公司,產(chǎn)地:安徽,批號:211001、211003、211206、220510、220602、220603),經(jīng)連云港市食品藥品檢驗檢測中心馬冬云主任中藥師鑒定為桑科植物桑(Morus alba L.)的干燥根皮。
2 方法與結(jié)果
2.1 溶液的制備
2.1.1 對照品溶液 分別取桑皮苷A、綠原酸、單寧酸、桑根酮D、桑酮G、桑根酮C 對照品適量,精密稱定,置于同一50 mL 量瓶中,加甲醇適量,經(jīng)超聲振蕩(頻率:50 kHz,功率:200 W)溶解后,用甲醇稀釋成桑皮苷A、綠原酸、單寧酸、桑根酮D、桑酮G、桑根酮C 的質(zhì)量濃度依次為1618、1609、1252、1642、1593、1549 μg·mL-1的混合對照品儲備溶液。精密吸取該儲備溶液1 mL,置于10 mL 量瓶中,用50%甲醇稀釋至刻度,搖勻,即得混合對照品溶液。
2.1.2 供試品溶液 取經(jīng)干燥后粉碎的桑白皮粉末約2.0 g,精密稱定,置于圓底燒瓶中,加入超純水約50 mL,回流提取2 h,過濾,濾渣繼續(xù)提取2 h,合并濾液,濃縮至10 mL,用0.45 μm 微孔濾膜濾過,取續(xù)濾液,即得。
2.2 色譜條件
色譜柱:Phenomenex Gemini C18(250 mm ×4.6 mm,5 μm);流動相:A 相為乙腈,B 相為0.2%甲酸水溶液,梯度洗脫(0~10.0 min,5.0%A→35.0%A;10.0~20.0 min,35.0%A →50.0%A;20.0~30.0 min,50.0%A;30.0~40.0 min,50.0%A →70.0%A;40.0~42.0 min,70.0%A→5.0%A;42.0~50.0 min,5.0%A);檢測波長:270 nm(用于單寧酸、桑根酮D、桑酮G、桑根酮C)、320 nm(用于桑皮苷A、綠原酸);柱溫:30℃;流速:1.0 mL·min-1;進樣量:20 μL。
結(jié)果表明,理論板數(shù)按桑皮苷A、綠原酸、單寧酸、桑根酮D、桑酮G、桑根酮C 計均大于8000,各待測成分峰與相鄰峰之間的分離度均大于1.5。色譜圖見圖1。
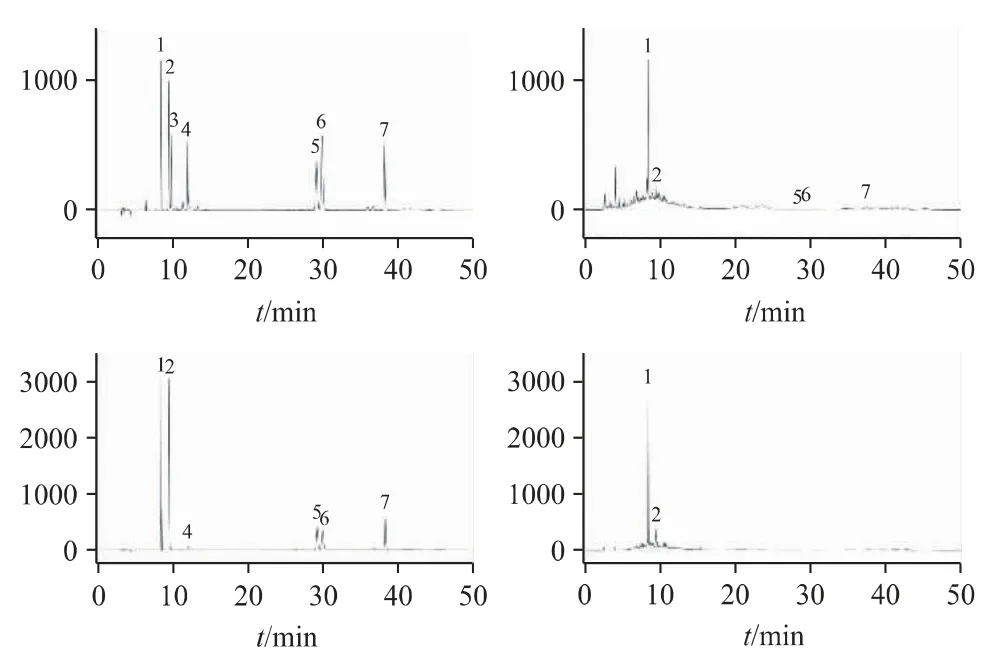
圖1 對照品和樣品的HPLC 色譜圖
2.3 方法學(xué)考察
2.3.1 線性關(guān)系考察 分別精密吸取“2.1.1”項下的混合對照品儲備溶液0.1、0.2、0.5、1、1、1、2mL,分別置于50、50、25、25、10、5、5 mL 量瓶中,用50%甲醇稀釋至刻度,搖勻,按“2.2”項下的色譜條件進樣測定,以各成分的質(zhì)量濃度(μg·mL-1)為橫坐標(biāo)(X)、峰面積為縱坐標(biāo)(Y)進行線性回歸,結(jié)果見表1。
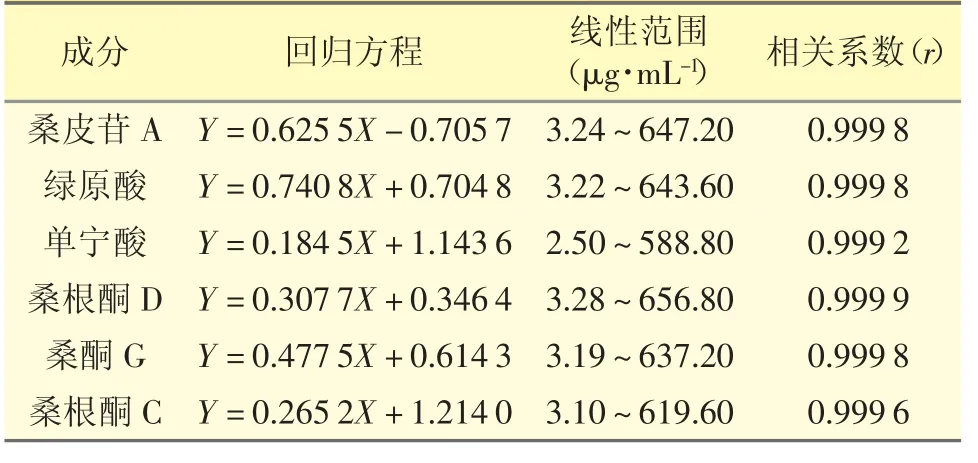
表1 6 個成分回歸方程、線性范圍及相關(guān)系數(shù)(n=7)
2.3.2 檢測限和定量限 將“2.1.1”項下的混合對照品溶液,用50%甲醇逐步稀釋至低濃度,以信噪比(S/N)3 作為檢測限(LOD),信噪比(S/N)10 作為定量限(LOQ),得出桑皮苷A、綠原酸、單寧酸、桑根酮D、桑酮G、桑根酮C 的檢測限分別為0.13、0.33、0.32、0.15、0.17、0.30 μg·mL-1,定量限分別為0.43、1.10、1.07、0.50、0.57、1.00 μg·mL-1。
2.3.3 進樣精密度試驗 取“2.1.1”項下制得的混合對照品溶液20 μL,按“2.2”項下的色譜條件重復(fù)進樣6次,記錄結(jié)果,計算得出桑皮苷A、綠原酸、單寧酸、桑根酮D、桑酮G、桑根酮C 峰面積的RSD(n=6)分別為0.41%、0.55%、0.83%、0.44%、0.25%、0.56%,表明儀器精密度良好。
2.3.4 重復(fù)性試驗 取同一批號(211001)的桑白皮樣品,分別按“2.1.2”項下的方法配制6 份供試品溶液,按“2.2”項下色譜條件平行測定6次,記錄色譜圖,用標(biāo)準(zhǔn)曲線法計算得出樣品中桑皮苷A、綠原酸、桑根酮D、桑酮G、桑根酮C 含量的RSD(n=6)分別為0.44%、0.70%、1.46%、1.39%、0.55%,結(jié)果表明本方法重復(fù)性良好。
2.3.5 加樣回收率試驗 取已知含量的桑白皮樣品(批號:211001)約2.0 g,共9份,精密稱定,分為3組,每組分別精密加入混合對照品儲備溶液(桑皮苷A、綠原酸、單寧酸、桑根酮D、桑酮G、桑根酮C的質(zhì)量濃度依次為1618、1609、1252、1642、1593、1549 μg·mL-1)0.8、1.6、2.4 mL,按“2.1.2”項下的方法制得供試品溶液,按“2.2”項下色譜條件進樣測定,記錄色譜圖,用標(biāo)準(zhǔn)曲線法計算回收率和RSD,結(jié)果上述6 個成分平均加樣回收率及相對標(biāo)準(zhǔn)偏差(RSD)分別為99.18%(0.45%)、99.06%(0.33%)、98.96%(0.59%)、98.76%(0.36%)、98.99%(0.47%)、98.90%(0.49%)。
2.3.6 耐用性試驗 選取3 種不同型號的色譜柱:Phenomenex Gemini C18柱(250 mm×4.6 mm,5 μm)、Agilent Eclipse Plus C18柱(250 mm×4.6 mm,5 μm)、Waters Supelco Discovery C18柱(250 mm×4.6 mm,5 μm),取“2.1.1”項下的混合對照品溶液,按“2.2”項下的色譜條件進樣測定并記錄結(jié)果。通過分析高效液相色譜圖,桑皮苷A、綠原酸、單寧酸、桑根酮D、桑酮G、桑根酮C 在3 種色譜柱上均能與其他組分得到滿意的分離度,樣品色譜圖中指標(biāo)成分的色譜峰與對照品中相應(yīng)峰在200~400 nm 波長范圍內(nèi)的UV 吸收光譜一致,表明方法耐用性良好。
2.3.7 穩(wěn)定性試驗 取同一批供試品溶液(批號:211001)適量,在室溫下放置0、2、4、8、12、24、48、72 h后,按“2.2”項下色譜條件進樣測定,結(jié)果24 h內(nèi)桑皮苷A、綠原酸、單寧酸、桑根酮D、桑酮G、桑根酮C 峰面積的RSD 分別為0.39%、0.68%(n=6),24 h 之后6 個成分的峰面積均減小30%以上,表明供試品溶液在24 h 內(nèi)穩(wěn)定性良好,超過24 h 供試品溶液中6 個成分均有較大程度降解,提示應(yīng)在24 h內(nèi)進樣,以保證測定結(jié)果的準(zhǔn)確。
2.4 含量測定
取收集到的6 批桑白皮樣品,按“2.1.2”項下方法制備供試品溶液,每批樣品平行制備3份,分別將混合對照品溶液和供試品溶液按“2.2”項下色譜條件進樣測定,記錄色譜圖。按外標(biāo)法計算含量,結(jié)果見表2。
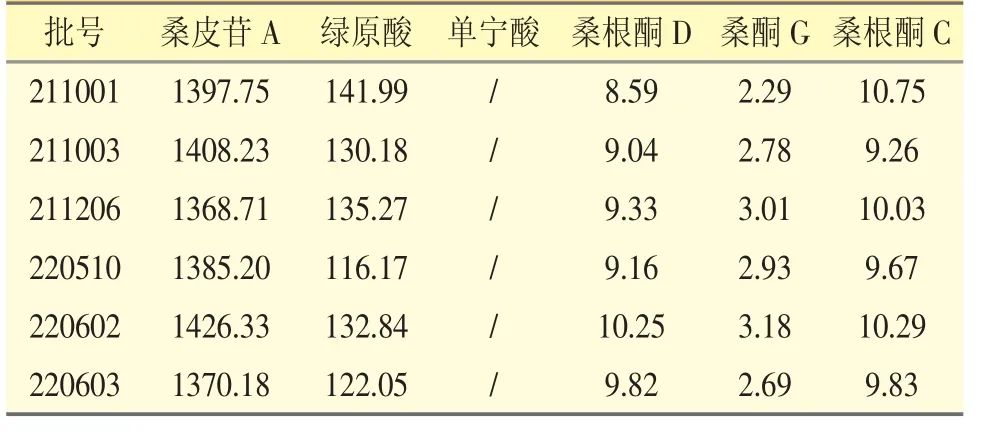
表2 桑白皮水提物中6 個成分的含量測定結(jié)果(μg·g-1,n=3)
3 討論
3.1 流動相的選擇
本試驗參考相關(guān)文獻[7,8],分別考察了甲醇、乙腈作為有機相,水、甲酸水溶液、磷酸鹽水溶液作為水相時桑皮苷A 等6 個指標(biāo)成分的色譜峰分離情況,發(fā)現(xiàn)當(dāng)選用甲醇-水作為流動相時,6 個成分的色譜峰對稱性差,理論塔板數(shù)低,且基線不平穩(wěn),影響檢測結(jié)果。當(dāng)有機相改為乙腈時,雖然各峰之間分離度達(dá)到要求,但仍有有不同程度的拖尾現(xiàn)象。比較甲醇-0.2%甲酸、乙腈-0.2%甲酸、乙腈-0.2%磷酸氫二鉀水溶液,各成分色譜峰峰型均有所改善,但乙腈作為有機相時色譜峰對稱因子更小,而且為使各成分色譜峰分離度符合要求,選擇洗脫能力強的乙腈更有優(yōu)勢。在乙腈-磷酸鹽體系下,各成分色譜峰均有良好的峰型;但從環(huán)保的角度考慮,還是選用0.2%甲酸水作為水相。由于各指標(biāo)成分極性有較大差異,故選擇梯度洗脫方式,從而縮短分析時間,提高分離效果。
3.2 檢測波長的選擇
按照上述確定的流動相,將混合對照品溶液進樣測定,采用二極管陣列檢測器,在波長200~400 nm范圍內(nèi)對待測分析物進行全掃描,結(jié)果如圖2 所示,桑皮苷A、綠原酸在波長216、325 nm 處有最大吸收,單寧酸在波長216、278 nm 處有最大吸收,桑根酮C、桑根酮D 在波長282、310 nm 處有最大吸收,桑酮G 在波長262、317 nm 處有最大吸收,綜合考慮末端吸收、檢測靈敏度和其他干擾因素,最終確定采用雙波長檢測法,在波長270 nm 處檢測單寧酸、桑根酮D、桑酮G、桑根酮C,在波長320 nm 處檢測桑皮苷A、綠原酸。
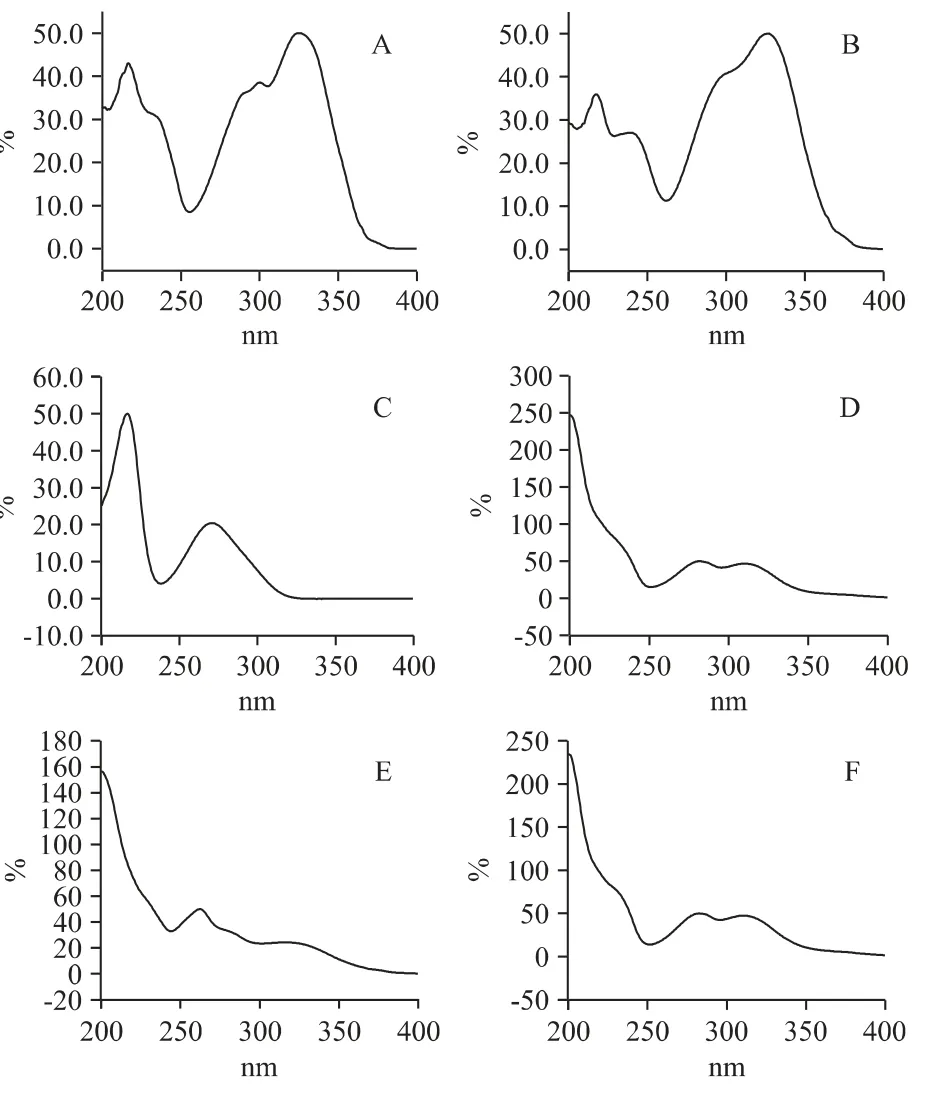
圖2 6 個成分的光譜圖
4 小結(jié)
建立的HPLC 雙波長法可同時測定桑白皮水提物中桑皮苷A 等6 個成分的含量,分析時間在40min內(nèi),方法準(zhǔn)確度、進樣精密度、重復(fù)性及耐用性均符合分析方法驗證要求,為定量分析桑白皮水提物中的主要成分提供了可靠的依據(jù)。同時對桑白皮水提物進行了穩(wěn)定性研究,表明桑白皮水提物在24 h 內(nèi)穩(wěn)定性良好,在超過24 h 供試品中6 個成分均有較大程度降解,為桑白皮水提物在化妝品領(lǐng)域的開發(fā)應(yīng)用提供了一定參考。
