CFHR3基因異常的非典型溶血尿毒綜合征1例并文獻復習
2022-01-27 08:18:32李天高向瑩萬單華陳虹
安徽醫藥 2022年2期
關鍵詞:途徑
李天,高向瑩,萬單華,陳虹
溶血尿毒綜合征(hemolytic uremic syndrome,HUS)是由多種病因引起的血栓性微血管病(thrombotic microangiopathy,TMA),以血小板減少、微血管性溶血性貧血及急性腎功能衰竭為主要表現的三聯征[1-2]。本病好發于嬰幼兒及學齡兒童,是引起小兒急性腎衰竭的常見原因之一。根據HUS的發病誘因,將其分為典型HUS和非典型HUS(atypical hemolytic uremic syndrome,aHUS)。典型HUS又稱腹瀉后HUS,因為大部分HUS繼發于產志賀樣毒素的細菌感染,起初aHUS被定義為非腹瀉相關型HUS。根據目前最新國際分類,aHUS特指補體替代途徑調控異常所引起的血管內皮功能失調和微血管血栓形成。aHUS在兒童中的發病率為0.10/10萬~0.11/10萬[3-4],其臨床表現在不同個體之間差異較大,多表現為進行性、破壞性進展,病情易反復、預后差,aHUS急性期病死率約為25%,約50%的病例進展為終末期腎病(end-stage renal failure,ESRF)[5]。
1 臨床資料
男,1歲,主因“間斷發熱10 d,面色發黃伴嗜睡半天”于2019年7月9日入院。于入院前10 d“受涼”后出現發熱,熱峰39.5℃,無驚厥,無嗜睡,無惡心、嘔吐,無腹瀉,無哭鬧,當地診所診斷為“急性上呼吸道感染”,給予對癥治療7 d,效果不佳,病兒仍反復發熱,遂就診于當地縣醫院,查血常規:白細胞11.96×109/L,淋巴細胞百分比60.8%,中性粒細胞百分比25.4%,紅細胞5.61×1012/L,血紅蛋白139 g/L,血小板108×109/L。給予對癥治療3 d,病兒體溫較前減低,熱峰37.8℃。于入院前半天,出現面色發黃,伴乏力、嗜睡,納奶明顯減少,余無特殊不適,再次就診于當地縣醫院,查血常規+C反應蛋白(CRP):白細胞28.99×109/L,淋巴細胞百分比0.397,中性粒細胞百分比0.548,紅細胞4.11×1012/L,血紅蛋白139 g/L,血小板102×109/L,CRP14.07 mg/L。天冬氨酸氨基轉移酶132 U/L,丙氨酸氨基轉移酶1 222 U/L,總膽紅素 229 μmol/L,直接膽紅素 149.1 μmol/L,間接膽紅素74.9 μmol/L。以發熱待查、肝功能異常就診于蘭州大學第一醫院。自發病以來,病兒大便色黃,量可,小便量無明顯減少,色黃。既往無特殊病史。無毒物及化學物質接觸史。無家族遺傳病史。
入院查體:嗜睡狀態,皮膚黏膜輕度黃染,無皮疹及出血點。雙肺呼吸音粗,可聞及濕性啰音。腹膨隆,肝臟肋下3 cm,劍下2 cm,脾臟肋下2 cm。雙側膝腱反射活躍,雙側巴氏征陽性,雙側克氏征陽性,雙側踝陣攣陽性。
輔助檢查:7月9日(入院第1天)查血常規+CRP、血生化(表1,表2);尿常規:葡萄糖±,余正常;糞便常規:正常;降鈣素原:2.220 ng/mL;血氨:139 μmol/L;凝血功能:抗凝血酶(AT)19%,凝血酶原時間(PT)55.4 s,凝血酶原活動度(PTA)13%,血漿凝血酶原比值(PTR)5.04,國際標準化比值(INR)4.96,纖維蛋白原含量(FIB)0.43 g/L,活化部分凝血活酶時間(APTT)76.9 s,凝血酶時間28.3 s,D-二聚體定量(D-D)34.83 μg/mL,纖維蛋白原降解產物(FDP)103.81 μg/mL;免疫球蛋白+補體:IgG 12.6 g/L,IgA 1.23 g/L,IgM 1.34 g/L,C3 0.31 g/L,C4 0.05 g/L;溶血篩查:間接抗人球蛋白試驗陽性;自身抗體:抗核抗體(ANA1)陽性,抗中性粒細胞質抗體(CANCA)陽性。胸部CT:右肺上葉胸膜下小斑片,炎癥可能。
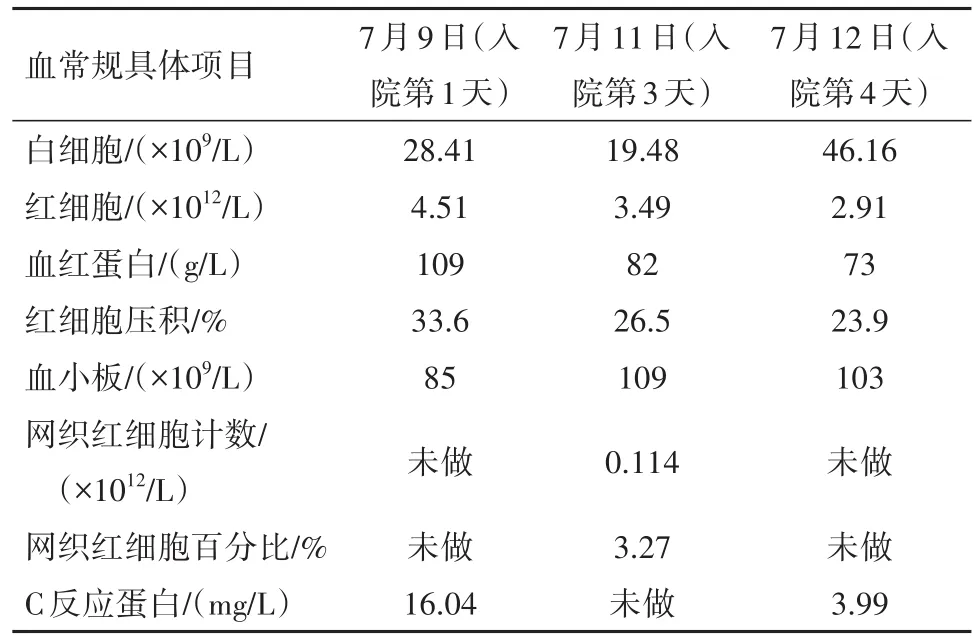
表1 病兒血常規+C反應蛋白結果變化趨勢
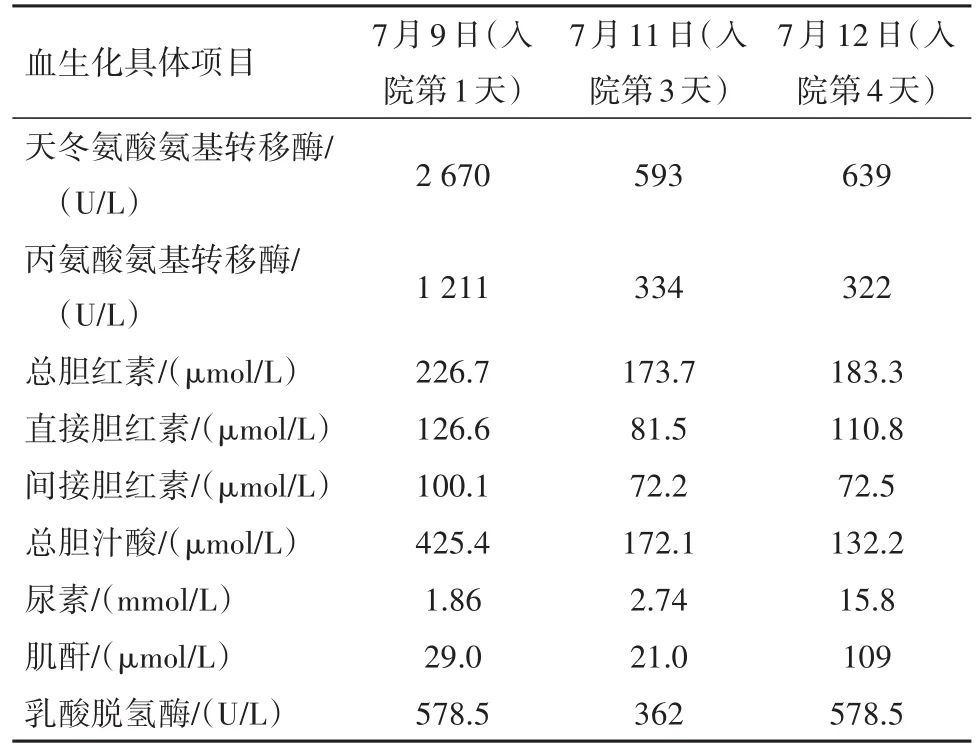
表2 病兒血生化結果變化趨勢
診療經過:病兒于7月9日入院后給予輸注血漿及凝血酶原復合物改善凝血功能、保肝、抗感染、連續性床旁血液凈化等對癥治療。7月10日(入院第2天)病兒尿色較前加深,尿量尚可,約4.07 mL·kg-1·h-1,有特殊氣味,呈淺昏迷狀態,抽搐3次,給予鎮靜對癥處理,伴消化道出血癥狀,給予禁食水、止血對癥處理,癥狀緩解,同時行第一次血漿置換治療,考慮病兒病情進展快,程度較重,向家屬交代病情,征得家屬同意后完善基因檢測。7月11日(入院第3天)病兒尿色呈深黃色,尿量可,約3.62 mL·kg-1·h-1,仍呈淺昏迷狀態,伴間斷抽搐,繼續予以連續性血液凈化及對癥處理,同時復查血常規+網織、血生化(表1,表2);尿常規:正常。7月12日(入院第4天)病兒尿色呈金黃色,尿量較前明顯減少,約2.37 mL·kg-1·h-1,仍處于淺昏迷狀態,仍有間斷抽搐,暫繼續連續性血液凈化及輸注紅細胞糾貧血等處理,余治療方案暫同前,再次復查血常規+CRP、血生化(表1,表2)。于7月12日傍晚病兒出現神志昏迷、嘆息樣呼吸,立即給予氣管插管同時連接呼吸機輔助呼吸,病兒病情逐漸轉平穩。約30 min后病兒再次出現病情變化,最終經積極搶救后醫治無效死亡。
2 結果
通過對先證者全外顯子組測序檢測,采用芯片捕獲高通量測序,其基因學檢查示:全外顯子組測序檢測發現CFHR3基因的1個變異,關聯疾病為非典型溶血尿毒綜合征,易感1型(OMIM:235400)。致病基因染色體位置為:chr1:196744016-196749103;核酸改變(外顯子號):1oss1(EXON:1-3);氨基酸改變(變體號):5K。本實驗將正常對照樣本與先證者及家系樣本進行同組熒光定量PCR,以ALB基因為內參基因,對目標基因CFHR3的1-3外顯子的拷貝數進行檢測(用熒光定量PCR法)。基因變異驗證結果:該病兒為先證者,先證者及先證者之母CFHR3基因的1-3外顯子的拷貝數與正常對照的比值約為0.5,提示先證者及先證者之母CFHR3基因的1-3外顯子存在雜合缺失;先證者之父CFHR3基因的1-3外顯子的拷貝數與正常對照的比值約為1.0,提示先證者之父CFHR3基因的1-3外顯子拷貝數正常。
3 討論
非典型溶血尿毒綜合征是一種復雜的、多基因補體介導的疾病,其特指補體替代途徑調控異常所致的血管內皮功能失調和血管微血栓形成(thrombotic microangiopathy,TMA)。其發病機制是由于補體替代途徑中的基因異常引起的,導致補體系統的過度激活和微血管血栓形成。補體通路的異常可能以補體關鍵基因突變或針對特定補體因子的自身抗體的形式出現[6]。
補體是先天免疫反應的一部分,幫助宿主細胞通過三種不同的途徑清除病原體,即:經典激活途徑、甘露聚糖結合凝集素(MBL)途徑和旁路激活途徑[7]。這3條途徑最終聚合產生C3轉化酶(C3bBb),其啟動膜攻擊復合物(membrane attack complex MAC)C5-9的形成。補體替代途徑在缺乏抗體的情況下激活先天免疫系統。在這一途徑中,C3b在遇到異物表面后被激活并與B因子結合,創建C3轉換酶(C3bBb)。然后C3轉化酶招募更多的C3b沉積在細胞膜上,生成C5轉化酶,負責MAC的形成和隨后的細胞死亡。替代途徑具有內建的調節因子,可以抑制未抑制的C3b沉積和對正常細胞的補體破壞。FH和FI調節C3轉化酶的形成。FI通過將C3b裂解為片段,而FH與C3b結合,作為FI裂解C3b的輔助因子,阻止C3轉化酶的形成,血栓調節蛋白降解C3a和C5a這些調節劑的任何缺陷都會導致補體通路的過度激活[8]。在非溶血尿毒綜合征中,替代途徑失去控制,導致膜攻擊復合物(MAC)的形成,以及多種致內皮損傷、血小板活化、炎癥反應、血栓、血小板減少、貧血和腎功能不全等病理特征的過敏性休克素的形成。C3裂解后,C3b與B因子結合,B因子又被D因子裂解為Bb因子。C3Bb復合物也被稱為C3轉化酶,形成了一個擴增環并產生更多的C3b。新的C3b片段與C3轉換酶結合,形成C5轉換酶。然后,C5裂解為C5a(炎性顆粒)和C5b。c5b最終與C6、C7、C8、C9結合形成MAC。這種級聯由多種蛋白調控,如因子I(CFI)及其輔助因子、因子H(CFH)、血栓調節蛋白和膜輔助因子蛋白(MCP)[9]。
在aHUS中檢測到大約一半的基因突變位于CFH中[10],可見CFH是調節旁路途經中最重要的蛋白質。首先,CFH不僅可以和CFB競爭與C3b的結合,從而阻止C3轉化酶的形成[11],而且可以將已經形成的C3轉化酶滅活[12];其次,在CFI降解C3b的過程中,CFH是必須的組分[13];另外H因子相關蛋白(CFHR1~5),在抑制C5轉化酶、MAC的組裝及插入以及加強H因子作為輔助因子的活性方面有重要作用[14-15]。由此可見CFH/CFHR蛋白家族在旁路途經的調節中處于核心地位,若該調節機制中單一或多個調節因子缺乏或被抑制,將會出現C3b滅活減少,形成大量C5轉化酶,最后導致MAC大量形成,造成自身組織器官損傷,從而導致疾病的發生。目前發現與aHUS相關的基因突變包括C3、C4、C5、CFH、CFB、CFI、MCP、CFHR1、CFHR3、CFHR5、THBD、PLG、DGKE[16]。在 aHUS病人中最常見的基因異常主要有CFHR1缺失、CFHR1/CFHR3聯合缺失、CFHR5 突變等[17]。26.5% 的病人有 CFHR3 和CFHR1缺失[18]。本病例中基因檢測結果提示為CFHR3基因突變所致。隨著基因檢測技術的發展,以及在該領域的應用,更多aHUS相關的補體旁路途徑突變正陸續被檢出和報道。此外,aHUS導致的腎衰竭可直接由MAC損傷和/或由血栓性或非血栓性狹窄引起的缺血性損傷引起。血栓、狹窄小動脈和毛細血管中剪切應力異常,血小板在血栓形成中消耗,可致血小板減少及微血管病變性溶血性貧血形成[19]。
Bernabéu-Herrero等[20]報道 CFHR3c.721T 變體在FHR-3的241位氨基酸處產生絲氨酸到脯氨酸的變化,與aHUS的風險增加相關。Zipfel等[21]在一項研究中發現CFHR1/CFHR3缺陷的血漿具有降低的保護活性,并且表明CFHR1和/或CFHR3的缺失有助于細胞和組織表面上補體活化的缺陷調節。報道了CFHR1和CFHR3的雜合和純合缺失通過CFH下游的非等位同源重組事件與aHUS風險增加相關。缺乏CFHR1/CFHR3的aHUS病人的特征是發病時年齡相對較小(1~21歲)。
IgA腎病是一種自身免疫性疾病,其中含有的IgA1-免疫復合物引發腎小球損傷。朱莉等[22]的研究確定了CFH,CFHR3和CFHR1變體與IgA腎病病人循環CFH水平和系膜C3的沉積有顯著的相關性。這些關聯表明這些變體對IgAN中補體激活的調節作用,并為CFH,CFHR3和CFHR1變體引起的IgAN易感性提供了可能的遺傳機制。然而關于補體活化在IgA腎病發病的確切機制我們的理解仍然是有限的。Fritsche等[23]研究發現C5a是有效的過敏毒素,可將嗜中性粒細胞吸引到感染一側,并引發和增強炎癥反應。CFHR3和CFHR1減少了C5a的生成,因此阻斷了補體介導的嗜中性粒細胞趨化性。故CFHR3和CFHR1也具有抗炎作用。
本病例中病兒有發熱、昏迷、抽搐、溶血、血小板減少等特點,符合非典型溶血尿毒綜合征的表征。病兒通過行基因測序發現CFHR3基因外顯子區域出現一處雜合基因突變導致編碼的氨基酸發生變異,且本文報道的變異體在既往國內外文獻中關于aHUS病人CFHR3遺傳變異類型中尚未提及。目前由于CFHR3在HUS和其他腎臟疾病中的生物作用尚不明確,其基因序列變異的意義仍有待進一步研究,同時作為臨床工作者更應重視基因篩查對疾病診治的臨床意義。
猜你喜歡
語數外學習·高中版中旬(2023年2期)2023-05-10 13:26:53
語數外學習·高中版中旬(2022年5期)2022-07-13 20:47:51
中學生數理化·七年級數學人教版(2019年10期)2019-11-25 07:33:58
中學生數理化·高一版(2018年9期)2018-10-09 06:46:50
湖南教育·C版(2018年3期)2018-06-05 16:54:36
中學生百科·大語文(2017年10期)2017-11-04 06:56:38
中國衛生(2016年3期)2016-11-12 13:23:26
公民與法治(2016年22期)2016-05-17 04:20:13
中國衛生(2014年12期)2014-11-12 13:12:52
癌變·畸變·突變(2014年6期)2014-02-27 06:15:03
