零維銻基有機-無機雜化氯化物的自陷態激子發光及其發光二極管
2022-01-23 13:52:20蔡培慶滕嶸馭占宇鑫王祥夫司俊杰劉祖剛
發光學報 2022年1期
蔡培慶, 滕嶸馭, 張 帝, 王 淞, 占宇鑫, 王祥夫, 司俊杰, 姚 鑫, 艾 琦, 劉祖剛
(1.中國計量大學 光學與電子科技學院, 浙江 杭州 310000;2. 南京郵電大學 電子與光學工程學院, 微電子學院, 江蘇 南京 210023)
1 引 言
鉛基金屬鹵化物鈣鈦礦材料由于具有優異的光電性能,近年來引起了廣大研究者的關注[1-5]。然而,其中存在的可溶性重金屬鉛元素對人體健康存在隱患,因此無鉛金屬鹵化物材料逐漸成為研究熱點。目前,人們已經開發出多種具有較高能量轉化效率的全無機無鉛金屬鹵化物材料和有機-無機雜化金屬鹵化物光電材料,并研究了這些材料在紫外光探測[6]、X射線成像[7]、太陽能電池[8]、照明與顯示[9]等領域的應用。
在無鉛金屬鹵化物材料中,零維金屬鹵化物材料具有多變的空間幾何特性和獨特的電子結構,因此,關于該類化合物的光致發光研究已有大量的文獻見諸報道,華南理工大學夏志國教授近期綜述了該領域的進展[10-11]。零維金屬鹵化物發光材料的發光來源主要有兩大類。一類是孤立發光中心的發光,如摻雜于其中的稀土離子或者過渡金屬離子;另一類是由零維金屬鹵化物的晶格受到光照或者偏壓等誘導產生變形和極化,由此產生的束縛自陷態激子(Self-trapped exciton, STE)發光,屬于復合發光的范疇。近期,該類STE發光現象引起了大量研究者的關注[12-14]。零維金屬鹵化物材料的自陷態激子發光一般有如下特點:(1)由于空間位置的限域特性和三維空間內較強的介電屏蔽作用,激子的擴散距離往往較短,體現出類似于弗蘭克爾激子和空間轉移激子的特性;(2)激發態下的激子由于受晶格的束縛,電子-晶格耦合作用較強,具有較大的弗蘭克-康登因子(Frank-Condon-factor),發光峰型較寬,斯托克斯位移一般較大,光子循環作用較弱,量子產率較高;(3)由于自陷態激子受零維金屬鹵化物中存在的鹵素和金屬的重原子效應影響,自旋-軌道耦合作用較為明顯,因此一般呈現出微秒級別的三重態自陷激子磷光壽命。然而,目前關于自陷態激子電致發光(Electroluminescence,EL)現象的報道較少,主要有CsPbI3體系[15-17]、Cs2AgInCl6體系[18]、Cs3Cu2I5/CsCu2I3體系[19-22],以及本課題組報道的二維鈣鈦礦體系[23]等。因此,開拓出新型基于自陷態激子發光的電致發光器件對金屬鹵化物光電器件的無鉛化研究非常重要。
芳香膦(氧)基團由于結構上打破共軛,可以同時調控電子效應和空間效應,因此非常適合用于構建綜合性能優異的電致發光材料和器件。黑龍江大學許輝教授在該方面有綜合性的評述[24];2017年,福建物構所陳忠寧研究員報道了基于四苯基膦溴化錳的綠光電致發光器件,最高外量子效率可達10.49%[25];2018年,Ma等報道了光致發光量子效率接近100%的四苯基膦氯化銻橙光材料[26]。因此,通過芳香膦基無鉛金屬配合物獲得高效電致發光器件是實現無鉛化器件的可行途徑之一。
本文通過反溶劑結晶的方法,合成出具有較高發光強度的零維四苯基膦氯化銻[(C6H5)4P]2SbCl5(以下縮寫為TPP2SbCl5)材料,對其發光性質進行了研究。并通過溶液加工和真空鍍膜等工藝,成功制備出基于自陷態激子的暖白光電致發光器件。
2 實 驗
2.1 樣品制備
粉末樣品的制備:首先將四苯基氯化膦(C6H5)4PCl(阿拉丁,98%)和三氯化銻SbCl3(阿拉丁,99.98%)按2∶1的量比稱取樣品,溶于二甲基亞砜DMSO(阿法埃莎,99.9%)溶液中;然后將裝有DMSO溶液的5 mL小瓶置于盛有10 mL乙醚(國藥,分析純)的50 mL燒杯中,將燒杯的開口塑封;通過乙醚的反溶劑作用,靜置一周左右可析出大量白色沉淀,將產生的白色沉淀離心并置于130 ℃的真空干燥箱內干燥過夜,所得粉末產物即為最終產物TPP2SbCl5。
薄膜樣品的制備:在充滿氮氣的手套箱中使用DMSO和N,N-二甲基甲酰胺DMF(阿法埃莎,99.9%)溶解上述獲得的白色產物,然后將溶液旋涂在潔凈的石英片上,將石英片置于120 ℃的熱臺上退火,獲得薄膜樣品。
2.2 器件制備
將ITO玻璃基片依次使用洗滌劑Decon 90(英國迪康)、丙酮(國藥,分析純)、超純水、異丙醇(國藥,分析純)等超聲清洗,氮氣吹干并用等離子體清洗機處理后備用。
在空氣氛圍中,以3 000 r/min的轉速旋涂空穴注入層聚3,4-乙烯二氧噻吩/聚苯乙烯磺酸鹽PEDOT∶PSS(西安寶萊特)于ITO玻璃基片上,110 ℃退火10 min,然后將基片轉移至氮氣手套箱內,再次旋涂空穴傳輸層聚[雙(4-苯基)(4-丁基苯基)胺]Poly-TPD(西安寶萊特,Mn≥60 000)的氯苯(阿法埃莎,99.9%)溶液,120 ℃退火10 min。然后在基片上旋涂含有TPP2SbCl5的DMSO溶液,并經過120 ℃退火30 min處理。將處理后的基片轉移至真空蒸鍍鍍膜機中,在真空度降低至5.33×10-4Pa (4×10-6torr)后,分別蒸鍍電子傳輸層材料1,3,5-三(1-苯基-1H-苯并咪唑-2-基)苯TPBi(西安寶萊特,99.9%)、電子注入層LiF(中諾新材,99.99%)和金屬Al(中諾新材,99.999%)電極,最后使用紫外固化膠(樂泰3492)和玻璃蓋板完成對器件的封裝,用于下一步的器件測試。
2.3 表征手段
使用德國Bruker D8型X射線衍射儀測量粉末和薄膜的物相信息;利用愛丁堡穩態瞬態熒光光譜儀FLS1000測量樣品的光譜數據;穩態光源選用的是歐司朗除臭氧的450 W氙燈,穩態激發光譜(Photoluminescence excitation,PLE)和發射光譜(Photoluminescence emission,PL)數據分別經過愛丁堡FLS1000內置的氙燈譜線參考文件和濱松R928P型光電倍增管(Photomultiplier tube,PMT)矯正文件進行矯正;瞬態光譜測試使用愛丁堡375 nm皮秒脈沖激光器作為光源,利用單光子計數技術測量熒光衰減曲線。通過東方晨景NCV30-2W-H型液氮低溫恒溫器控制樣品的測試溫度。使用耐馳STA449F3同步熱分析儀進行熱重分析,使用臺階儀(美國KLA Alpha-Step D-500)測試器件各功能層的厚度。利用Thermo ESCALAB XI光電子能譜儀測量紫外光電子能譜(Ultroviolet photoelectron spectrometer,UPS)。采用Keithley 2400電流-電壓源表、硅光電探頭和PR670光度計構成的測試系統測量電流(I)-電壓(V)-亮度曲線和外量子效率(External quantum efficiency,EQE)。
3 結果與討論
3.1 TPP2SbCl5的室溫光致發光和結構性質分析
圖1(a)是粉末樣品的發射光譜和激發光譜。激發光譜表明TPP2SbCl5可以在260~410 nm左右的紫外區域內被有效激發,光譜在紫外區的強度分布較為平坦,沒有明顯的銳線激子特征峰出現,激發峰邊在370 nm左右以后快速變陡,以上特征表明該物質具有基質晶格吸收特性。由于本實驗中使用的激發光源為除臭氧型氙燈,光源在高于260 nm以上的紫外區域內強度較弱,因此無法探測該物質在真空紫外區的激發情況,相關性能后期可以通過真空紫外光譜技術研究。另外,已有報道表明有機無機雜化鹵化物的自陷態激子可以被X射線等高能射線有效激發[27],因此,該類物質也可能有著良好的閃爍性能,相關性能有待進一步研究。
發射光譜顯示該物質的發射峰值在700 nm左右,為一寬帶發射,可以將其歸于自陷態激子的發射;發射光譜與激發光譜有著較大的斯托克斯位移,表明該物質的發光可以用強耦合模型的位型坐標圖去解釋。由于濱松R928P型PMT的探測范圍限制,800 nm以后的發射區域噪聲過大,因此超過800 nm的光譜區域并未掃譜。圖1(a)中插圖顯示,該粉末在日光照射下為一白色物質,表明其在可見光區沒有吸收;在365 nm的紫外燈激發下,該粉末發射出肉眼可見的明亮的橙紅光,但這與實測發射光譜峰值位于700 nm是不一致的。該現象可以解釋如下:由于該物質在500~900 nm左右均有非常強的寬帶發射,但人眼對680 nm以上的深紅光不敏感,所以人眼觀測到該物質的發光為橙紅光。另外,如圖1(a)中插圖所示,通過透過650 nm的長波通濾波片觀察,我們可以確認該物質存在深紅光區域的發射,這也意味著該物質在植物生長補光領域有著潛在的應用[28]。

圖1 (a)TPP2SbCl5粉末的激發/發射光譜,激發光譜的監測波長為700 nm,發射光譜的激發波長為365 nm,插圖從左到右分別為粉末樣品在日光下、在365 nm紫外燈激發下以及在365 nm紫外燈激發下通過650 nm長波通濾波片拍攝的照片;(b)TPP2SbCl5粉末和薄膜的發射光譜對比,插圖自上往下分別為在365 nm紫外燈激發下,使用DMSO、DMF旋涂的TPP2SbCl5薄膜,以及TPP2SbCl5粉末的熒光照片;TPP2SbCl5粉末和薄膜的XRD圖譜(c)和結構圖(d)。
如圖1(b)所示,TPP2SbCl5的粉末PL譜與使用DMSO、DMF溶液旋涂的薄膜PL譜相比,譜線有所不同。Ma等在制備該樣品時發現了類似的現象。他們認為,由于金屬鹵化物自陷激子的熒光性質強烈依賴于軟晶格的結構變化,這種自陷激子熒光發射的差異是由不同的殘留有機溶劑分子導致材料熱力學穩定相的轉變造成的。研究發現,快速結晶的小顆粒薄膜樣品為熱力學不穩定相,薄膜的熒光呈現黃色;而緩慢結晶的大塊樣品是熱力學穩定相,其熒光則紅移至紅色[26,29]。圖1(b)插圖的薄膜樣品圖片顯示,使用DMSO旋涂而成的薄膜較為光滑,表明晶體顆粒較小;而用DMF旋涂而成的薄膜較為粗糙,表明晶體顆粒較大。在365 nm紫外光激發下,DMSO和DMF作為溶劑旋涂的TPP2SbCl5薄膜發射峰值分別在635 nm和660 nm左右,粉末的發射峰值在705 nm左右,隨著顆粒的增大,光譜逐漸紅移。因此,我們的實驗結果與他們的結論符合得很好。

3.2 TPP2SbCl5的變溫光致發光分析
變溫熒光性質的分析可以探究TPP2SbCl5激發態的一些性質。圖2(a)是TPP2SbCl5粉末從80~470 K的變溫熒光光譜。可以看到,熒光強度在室溫以下溫區的變化不太明顯,但隨著溫度升高至高于室溫的溫區,熒光強度呈現大幅度下降的趨勢,這與有機分子在室溫以上的分子振動增強有關。圖2(c)的熱重分析表明,在80~470 K內,樣品的重量并沒有明顯的變化,因此熒光強度的降低不是由于樣品受熱分解造成的。圖2(b)是TPP2SbCl5粉末的變溫熒光壽命圖譜。在80~470 K的測試溫區內,該樣品的自陷激子態發光均呈現出單指數衰減,并隨著溫度升高至室溫左右,壽命開始大幅降低。上述現象表明,在這種零維限域結構的有機金屬鹵化物內,自陷態激子復合發光衰減是一個單分子過程,并且該過程受無輻射弛豫的影響較為明顯[30-32]。另外,無論是在液氮溫度或是470 K左右的高溫下,該物質的發光始終保持在時間較長的微秒量級(5.741~0.289 μs),說明該物質的發光源于自陷激子的三重態禁阻發射[33]。

圖2 TPP2SbCl5粉末的變溫熒光光譜(a)和熒光衰減壽命(b);(c)TPP2SbCl5粉末的熱重分析圖;(d)~(e)熒光積分強度和熒光衰減壽命隨著溫度變化的計算結果和擬合曲線;(f)強耦合作用下的STE發光位型坐標模型圖。
通過以上熒光光譜和衰減壽命的分析,我們認為該物質的發光特征符合黃昆用于解釋F色心發光行為的高溫強耦合無輻射躍遷理論[34],可用如圖2(f)所示的位型-坐標模型去解釋。根據該理論的推導結果,不同溫度下的熒光積分強度I(T)和熒光衰減壽命τ(T)隨著溫度T的變化符合如下公式[35]:
(1)
(2)
其中I0是絕對零度時的發光強度積分,A是前因子,τr是輻射躍遷壽命,τnr是非輻射躍遷壽命,ΔE是熱激活能,k是玻爾茲曼常數。圖2(d)、(e)是分別對I(T)和τ(T)隨著T的變化擬合獲得的結果。擬合獲得的ΔE值分別為608 meV和603 meV,兩種擬合方式獲得的ΔE誤差值較小,表明實驗與理論符合得非常好。與室溫相對應的熱能(~26 meV)相比,600 meV左右的熱激活能較大,因此這是零維TPP2SbCl5的自陷態激子在室溫下沒有熱解離并能保持接近100%熒光量子效率的原因之一[26]。
3.3 電致發光器件的構筑和發光分析
通過UPS測試和吸收光譜計算,我們確定了如圖3(c)所示的器件結構圖,并經過逐層和多次的正交參數調控和優化,制備出以TPP2SbCl5為發光層的電致發光EL器件,器件參數為ITO/PEDOT∶PSS(40 nm)/Poly-TPD(30 nm)/TPP2SbCl5(60 nm)/TPBi(50 nm)/LiF(1.5 nm)/Al(100 nm)。圖3(a)是該器件的I-V-L曲線,從圖中可以看出器件的啟亮電壓為3.2 V,在6 V電壓下,器件亮度達到126 cd/m2。圖3(b)展示了該器件外量子效率EQE與器件電流密度的函數曲線。數據結果表明,器件的最高EQE為0.39%。圖3(d)為Poly-TPD/TPP2SbCl5薄膜的歸一化PL光譜及器件EL的對比,可以看出器件的藍光發射來源于空穴傳輸層的Poly-TPD發光,橙光來源于TPP2SbCl5的自陷態激子發光。但是,TPP2SbCl5的EL光譜與PL光譜相比在高能區和低能區均變窄。我們經分析認為,影響峰型窄化的因素如下:(1)首先,從能級的角度來看,無論是光致發光還是電致發光,Kasha規則確保光子必須在最低激發態下發射,自陷態激子發生T2→S0輻射復合造成高能區一側譜線變寬的可能性很小;(2)振動輔助的輻射躍遷(Vibration assisted radiative transition)幾率降低可以導致譜線窄化。在PL過程和EL過程中,一些在PL過程中允許的振動輔助躍遷在EL過程中很可能是禁阻的,載流子在STE T1和S0的振動能級上復合的幾率存在差異;在偏壓作用下,分子的剛性增強也可能會導致振動減弱,導致STE EL光譜窄化。

圖3 (a)器件的I-V-L曲線;(b)器件的EQE與電流密度函數曲線;(c)器件的能級結構圖;(d)Poly-TPD/TPP2SbCl5薄膜PL譜及器件EL譜比較;(e)不同電壓下的器件EL譜;(f)不同電壓下的CIE坐標,插圖為器件在6 V電壓下的照片。
不同電壓下的器件EL譜(圖3(e))表明,隨著電壓的增大,藍光部分逐漸增強,器件運行的視頻請見本文的補充文件。圖3(f)的Commission Internationale de L’Eclairage (CIE)1931圖更加明顯地顯示了不同電壓下色坐標的變化。從圖3(c)的器件結構能級圖中可以看出,該器件空穴傳輸層最低占據軌道(Lowest unoccupied molecular orbital,LUMO)能級與發光層LUMO能級差較小(0.16 eV),無法有效阻擋電子,而電子傳輸層最高占據軌道(Highest occupied molecular orbital,HOMO)能級和發光層HOMO能級的能級差較大(0.72 eV),阻擋空穴效果較好。因此,器件在工作過程中電子與空穴并未達到注入平衡。隨著電壓增大,更多的電子到達空穴傳輸層Poly-TPD處,導致Poly-TPD藍光增強[36]。另外,如圖3(f)插圖所示,器件在6 V正置偏壓下可發出明亮的暖白光,色坐標為(0.36,0.31),表明該器件在無鉛金屬鹵化物白光照明上有著潛在的應用。圖4為器件在較低電流密度20 mA/cm2下亮度隨時間變化的老化測試。其在初始亮度110 cd/cm2下的T90為5 min,器件的穩定性能有待進一步改善。
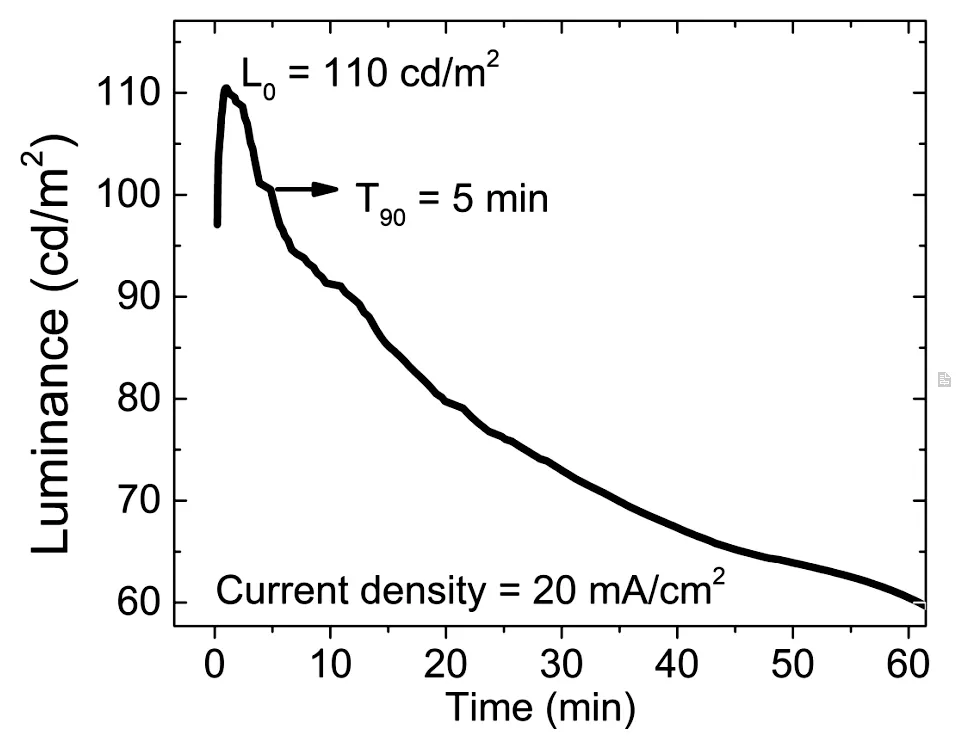
圖4 器件在20 mA/cm2電流密度下亮度隨時間的變化
4 結論與展望
本文成功制備了TPP2SbCl5發光材料和器件,并對其光致發光和電致發光性能進行了研究。結果表明,在紫外光激發下,TPP2SbCl5可以發出明亮的橙紅光,這種橙紅光源于零維限域作用下的自陷態激子三重態發光;變溫PL和衰減壽命研究表明該物質具有600 meV左右的熱激活能,抗熱猝滅性能較強。通過優化器件結構,引入Poly-TPD作為空穴傳輸層,獲得了在6 V偏壓下126 cd/m2的暖白光電致發光器件,但是由于TPP2SbCl5的成膜性能一般,器件的效率和壽命有待進一步改善。
在后續有關無鉛銻基STE電致發光的研究中仍需關注如下幾點:(1)STE電致發光無鉛材料的溶液法薄膜制備工藝有待進一步優化,雙源共蒸發薄膜制備工藝有待研究;(2)STE發光的斯托克斯位移較大,在電場作用下熱損耗不可避免,對器件的壽命影響較大,因此需要降低STE的斯托克斯位移;(3)STE結構上類似F色心,是一種激發態下的暫態局域化激子,存在飽和效應,因此需要調控晶體結構來提高自陷激子態密度,以提高單位電流密度下的復合效率;(4)一般STE的發光為三重態磷光,衰減壽命較長,對器件的正常工作是不利因素,因此,通過降低STE S1和T1能級的能級差,引入熱激活延遲熒光機制(Thermally activated delayed fluorescence,TADF),是有效提高STE電致發光的可能途徑。
本工作是對有機無機雜化零維無鉛金屬鹵化物的自陷態激子發光用于發光二極管的一次有益嘗試,有望進一步推動溶液法加工金屬鹵化合物電致發光器件的無鉛化進程。
本文專家審稿意見、作者回復信及補充文件的下載地址:http://cjl.lightpublishing.cn/thesisDetails#10.37188/CJL.20210318.
