群體基因組結構變異檢測工作流
2021-12-31 07:24:00曹舒淇劉詩琦
生物信息學 2021年4期
曹舒淇,劉詩琦,姜 濤
(哈爾濱工業大學 計算學部,哈爾濱150001)
基因組結構變異(Structural Variation,SV)是基因組上大尺度的核苷酸序列重排性變化,它包括長度大于50 bp的插入(INS)、缺失(DEL)、倒位(INV)、重復(DUP)、易位(BND)[1]。相關研究表明,平均每個人類個體上存在大約兩萬個結構變異[2],結構變異盡管相較于單核苷酸變異(SNV)、短插入缺失變異(INDEL)數量較少,但因其變異長度較大,因此對基因組上核苷酸序列的影響是最廣泛的[3]。結構變異會改變基因序列信息,進而影響轉錄過程,改變蛋白質空間結構,從而引發性狀與表型的改變[4]。此外,結構變異對基因表達調控[5]、種群多樣性[6]等方面有著重要影響,同時與以自閉癥[7]、阿爾茲海默癥[8]等為代表的許多疾病的引發有密切的關系。
結構變異會對人類遺傳、進化產生影響,形成個體之間的差異,影響種群的發展與演進。對于同一個群體,相當數量的結構變異對于群體中大部分個體是共享的,這些共享的結構變異可以有效對群體的特征與結構進行刻畫[9]。此外,在群體中仍存在個體特有的結構變異,這些個體特有的結構變異反映了個體獨有的特性,通過對特有結構變異以及個體表型的分析,能夠發掘結構變異與表型、疾病之間的重要關系[10]。
隨著國際千人基因組計劃的實施與推動[11-13],各國也紛紛啟動了本國的大規模人群基因組計劃[14-17],希望通過分析和構建本國、本民族的基因組變異圖譜,更加深入地解讀本國人群在遺傳、進化上的機理,為接下來開展的疾病診治、精準健康發展提供支撐。結構變異作為對基因序列影響最為廣泛的基因組變異類型,如何高效、精準的檢測群體結構變異已成為當前群體基因組研究中的核心。因此我們基于多層過濾的質量控制,多種算法的聯合檢測、多維度變異融合和校對,開發了一個高性能的群體結構變異檢測工作流,實現了群體基因組結構變異的全面、精準檢測。該工作流總體分為四個環節:基因組測序片段比對,單樣本基因組結構變異檢測,單樣本基因組結構變異融合以及群體基因組結構變異檢測(見圖1)。
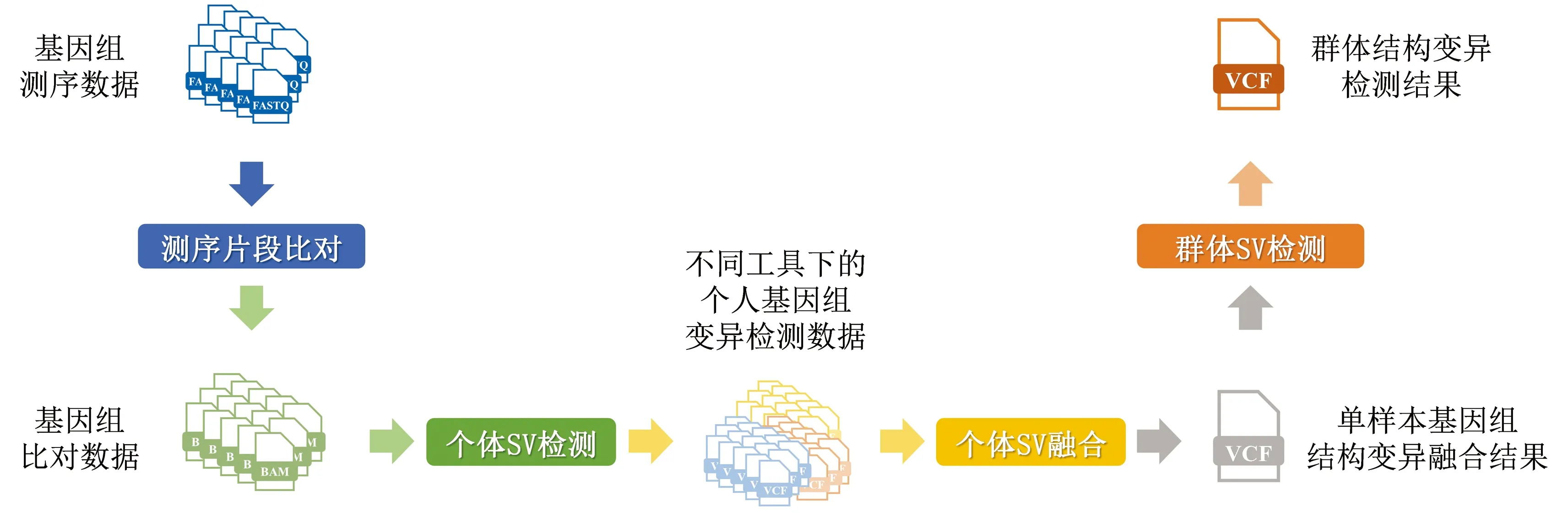
圖1 群體基因組結構變異檢測工作流Fig.1 Workflow of structural variation detection from population genomes
1 基因組測序片段比對
高通量基因組測序片段比對是基因組數據分析的首要環節,測序片段的比對的精度將對變異檢測、基因組拼接等下游分析產生重要的影響。因此,對基因組片段測序數據、片段比對數據等有效的質量控制,是保障以測序片段比對為基礎的基因組數據分析的關鍵。為此,本研究設計了多重質量控制與過濾的基因組測序片段比對工作流,該工作流主要包含以下步驟(見圖2)。

圖2 基因組測序片段比對流程Fig.2 Workflow of sequencing read alignment
(1)使用測序片段質量評價算法FastQC(https://github.com/s-andrews/FastQC)(v0.11.9,默認參數)對各樣本基因組測序數據進行質量控制,通過對測序片段中GC含量、重復性、堿基質量、片段長度分布等指標進行統計和閾值判定,若任意滿足:測序片段GC含量與理論分布偏差30%;測序片段重復度超過理論重復總量的50%;測序片段任意位置的堿基質量下四分位數低于5或中位數低于20;任意測序片段長度不足或長于150 bp,則將其認定為低質量測序樣本數據,并進行過濾處理。
(2)使用高通量測序片段比對算法BWA (https://github.com/lh3/bwa)(v0.7.17,默認參數),完成各測序樣本向參考基因組序列的比對。使用比對格式轉換算法Sambamba[18](v0.8.0,默認參數)對各樣本比對結果進行格式轉換和排序;使用測序重復片段標記算法Samblaster[19](v0.1.2,默認參數)對各樣本轉換后的比對文件進行重復標記;使用GATK (https://github.com/broadinstitute/gatk)(v4.2.0.0,默認參數)對測序片段比對中堿基質量校正形成最終的片段比對數據。
(3)使用測序片段比對質量評價算法Qualimap[20](v2.2.1,默認參數)對各樣本測序片段比對結果進行質量控制,通過對片段比對中測序覆蓋度(不低于30×測序深度)、片段重復性(不高于5%)、片段比對率(不低于95%)等指標進行統計和閾值判定,進一步過濾低質量測序樣本數據。
(4)使用DNA污染估計算法Verifybamid[21](v2.0.1,默認參數)計算各樣本中DNA污染程度,過濾高污染率(高于3%)測序樣本數據,形成最終用于下游分析的群體樣本集合。
多重質量控制與過濾的基因組測序片段比對工作流在完成各樣本測序片段比對任務的同時,將有效監控測序數據質量、比對數據質量、樣本污染情況等多重指標,為高質量基因組結構變異檢測奠定基礎。
2 單樣本基因組結構變異檢測
受限于高通量測序數據讀長與系統性測序誤差的限制,采用單一結構變異檢測工具識別各樣本基因組中的結構變異往往存在敏感性與準確性較低的問題,這將制約結構變異的檢測能力和向下游研究轉化的水平。針對這一問題,本研究采用三款當前性能最好的個體基因組結構變異檢測算法,全面挖掘各樣本基因組結構變異,主要步驟如下(見圖3)。

圖3 單樣本基因組結構變異檢測流程Fig.3 Workflow of structural variation detection from individual sample
(1)使用快速檢測基因組結構變異檢測算法Manta[22](v1.6.0,默認參數)識別各基因組中DEL變異、INS變異、INV變異、BND變異、DUP變異。
(2)使用簡化集成基因組結構變異檢測與基因分型算法Smoove(https://github.com/brentp/smoove)(v0.2.7,默認參數)識別各基因組中DEL變異、INS變異、INV變異、BND變異、DUP變異。
(3)使用拷貝數變異檢測算法CNVNator[23](v0.4.1,默認參數)識別各基因組中的拷貝數變異(CNV),并計算各基因組區域上測序覆蓋度信息。
分別使用Manta、Smoove、CNVNator三種結構變異檢測算法,將有效挖掘每個樣本基因組中多種類型結構變異,為融合形成群體基因組結構變異提供支撐。
3 單樣本基因組結構變異融合
群體基因組結構變異主要由各單樣本基因組結構變異融合產生。如何對來自不同樣本、不同檢測算法形成的結構變異進行融合,是當前產生高精度群體基因組結構變異的核心。為此,本研究分別對群體樣本中由同種檢測算法與不同檢測算法預測的結構變異分層次整合,從而產生最終群體結構變異候選位點,主要步驟如下(見圖4)。
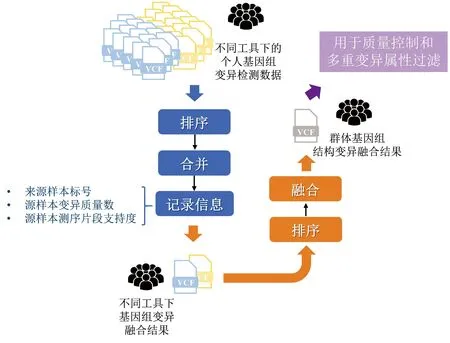
圖4 單樣本基因組結構變異融合流程Fig.4 Workflow of integration of individual structural variation
(1)分別對由Manta、Smoove檢測算法產生的結構變異按照基因組坐標進行排序,完成在相同檢測算法上不同樣本結構變異的融合。若相鄰兩個結構變異存在交疊,則將兩個變異合并為一個變異,直至所有變異均不存在交疊性。對于合并后的結構變異,分別記錄來源樣本標號,同時以累加方式累積各來源樣本在此變異上的變異質量數、測序片段支持度等信息。
(2)再次對由Manta、Smoove檢測算法產生的基因組結構變異融合結果進行排序,完成對不同檢測算法產生的融合結構變異數據的二次融合。
通過使用不同工具對各樣本基因組中的結構變異雙重融合,在充分保留各樣本基因組中潛在的結構變異的同時,在融合過程中記錄每個結構變異的樣本支持情況、變異質量情況等信息,為過濾低質量群體結構變異提供了保障。
4 群體基因組結構變異檢測
在完成單樣本基因組結構變異融合后,進行質量控制和多重變異屬性過濾,是完成高質量群體基因組結構變異檢測,繪制高精度人群基因組結構變異圖譜的核心。本研究實現這一目標主要采用如下3個步驟(見圖5)。
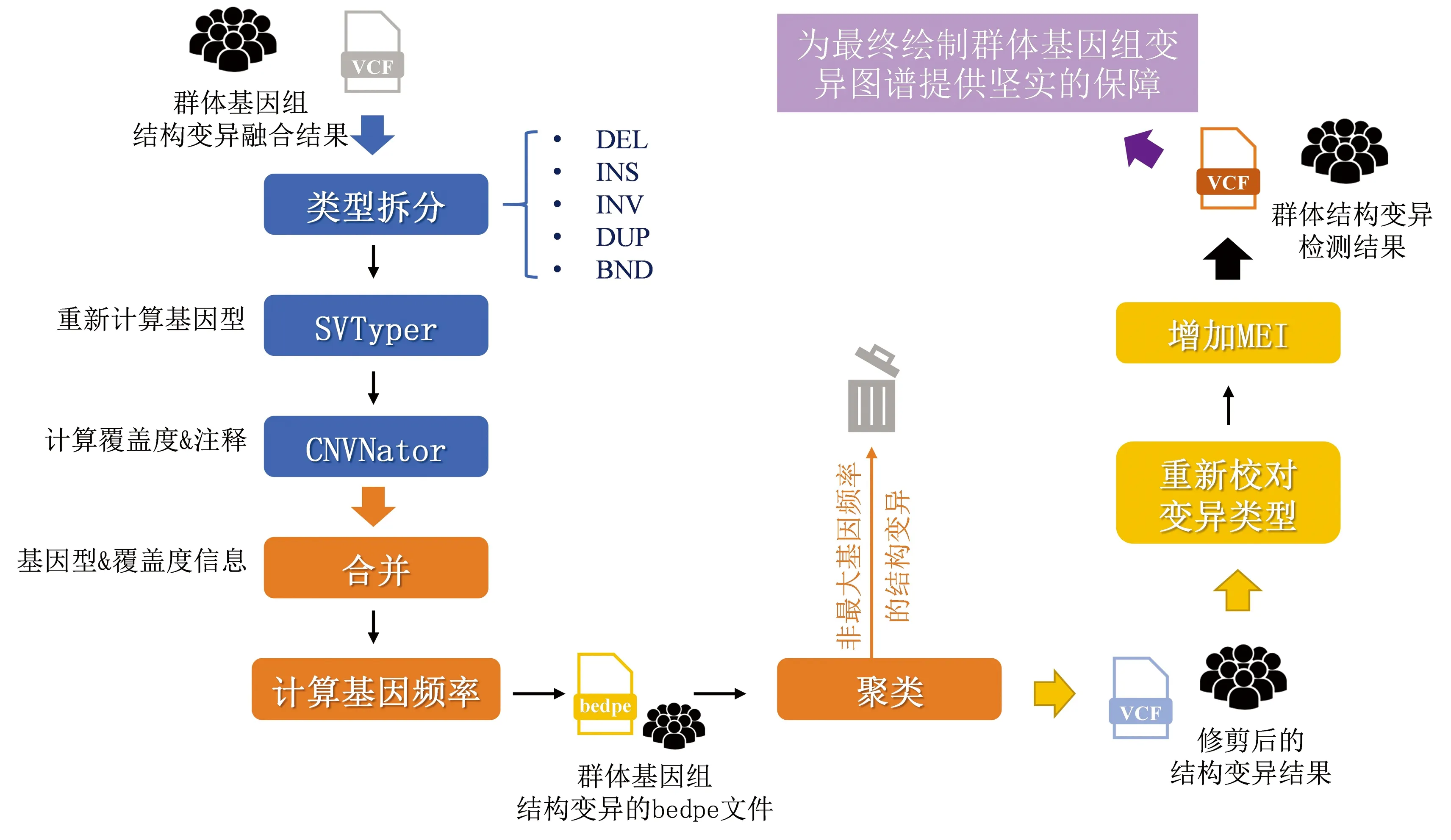
圖5 群體基因組結構變異檢測流程Fig.5 Workflow of structural variation detection from population genomes
(1)將融合后的基因組結構變異按照變異類型分別拆分為DEL、INS、INV、DUP、BND五種類型。使用基因組結構變異斷點基因型計算算法SVTyper[24](v0.7.1,默認參數),分別從單樣本層面對以上融合后的五種類型結構變異重新計算基因型。使用CNVNator計算的單樣本層面的測序覆蓋度信息對重新校準基因型信息的各結構變異進行注釋。
(2)對重新校正基因型和測序覆蓋度信息的各類型結構變異合并,計算各變異在人群中的變異頻率。將合并后群體結構變異檢測結果轉換為bedpe文件格式并排序,對存在變異區域交疊的結構變異進行聚類,保留聚類中具有最大變異頻率的結構變異,將聚類中其余結構變異修剪刪除。
(3)將經過修剪的結構變異集合重新轉換為vcf格式,依據測序覆蓋度信息和基因型一致性信息對各結構變異重新校對變異類型,新增移動元件變異(MEI)類型,形成最終的群體結構變異檢測結果。
經過對不同類型結構變異基因型的重新校正、過濾和變異類型校對,有效消減檢測形成的假陽性結構變異預測結果,最大限度反映群體中真實的結構變異位點、類型和變異頻率,為最終繪制群體基因組變異圖譜提供了堅實的保障。
5 結果與分析
為了驗證群體基因組結構變異檢測工作流的真實效果,本研究構建了由267個樣本組成的人群,使用Illumina高通量測序平臺對該人群樣本進行了30×高深度全基因組測序,并使用本研究提出的群體基因組結構變異檢測工作流對此267個樣本進行群體結構變異檢測(見表1),合計檢測出了96 202個結構變異,其中包括:11 697個DEL變異、18 385個INS變異、3 563個DUP變異、1 278個INV變異、2 007個MEI變異、59 272個BND變異。

表1 267樣本人群中結構變異檢測結果統計Table 1 Results of structural variation detection of 267 samples
在該267個樣本構成的人群中(見圖6),常見變異(AF≥0.05)占總體檢出變異的41%(39 086/96 202),低頻變異(0.05>AF≥0.01)占總體檢出變異的18%(17 554/96 202),罕見變異(0.01>AF)占總體檢出變異的41%(39 562/96 202)。值得關注的是,在DEL、DUP、INS、INV四種類型結構變異中,罕見變異的占比基本是均超過總體檢驗出變異的50%,相比之下,在MEI、BND兩種類型結構變異中,檢測出的常見變異數量是總體可檢測變異數量的主要占比。這些結果與過去開展的基因組計劃發現的結果相一致[25-26],說明本研究建立的群體基因組結構變異檢測工作流具有良好的檢測能力。

圖6 不同變異頻率中結構變異分布統計Fig.6 The distribution of structural variation among various allele frequencies
就每個樣本可檢測的結構變異而言,平均每個樣本可以檢測出18 388個結構變異,其中包含1 634個DEL變異、657個DUP變異、1 216個MEI變異、13 155個BND變異、1 521個INS變異、206個INV變異(見圖7)。受限于高通量測序技術中讀長的限制,基因組重復片段區域中的結構變異難以檢測和精確分型,其中僅可檢測到斷點連接關系的結構變異均歸結為BND變異,因此導致了每個樣本中包含了相當數量的BND變異。然而,僅獲取變異斷點連接關系,無法解析結構變異精準結構(如:是否為平衡變異,DNA變化方向等)將嚴重影響BND變異的可信度和準確性。經過對BND變異按照置信度進行過濾(見圖7),總計移除55 492個BND變異,僅保留3 780個高置信度BND變異(移除率93.62%)。平均每個樣本移除11 703個BND變異,僅保留1 451個高置信度BND變異(移除率88.97%)。

圖7 各樣本不同類型結構變異檢測數量分布和統計Fig.7 The quantitative distribution of various structural variation types among samples
此外,本研究還對群體基因組結構變異檢測工作流中變異融合與群體變異檢測兩個關鍵環節的計算開銷和內存使用進行了統計(見表2)。該工作流完成群體基因組結構變異檢測和融合兼容串行分析和并行分析兩種方式,其中串行計算方式需要約173.4 h,最大內存開銷30 GB,而采用并行計算方式僅需不足3 h,并維持最大30 GB的內存開銷。這一結果表明,對于大規模人群基因組結構變異檢測分析,在保持有限內存消耗的前提下,采用并行方式運行該工作流將顯著提升計算速度,為高效、快速的群體基因組結構變異檢測提供了保證。

表2 267樣本人群中結構變異檢測運行時間及內存統計Table 2 Time and memory cost of structural variation detection of 267 samples
6 結 論
1)本研究構建了一套高效、精準的群體基因組結構變異檢測工作流,該工作流通過多層過濾的質量控制,為高質量群體基因組結構變異檢測提供支撐。
2)該工作流通過使用多種高性能結構變異檢測算法,提高了結構變異檢測的準確性與敏感性,并通過雙重融合實現了群體結構變異候選位點的精準定位。
3)該工作流通過多維度重新校正結構變異候選位點的基因型與變異類別,進一步保障群體結構變異圖譜的高質量構建。
4)利用該工作流對由267個樣本組成的人群進行基因組結構變異檢測,結果表明該工作流具有良好、快速、高效的檢測能力。通過并行分析策略在控制內存消耗的基礎上,提高了工作流的計算速度,為大規模群體基因組研究提供了可能。
猜你喜歡
今日農業(2021年19期)2022-01-12 06:16:36
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中老年保健(2021年11期)2021-08-22 03:15:44
哲學評論(2021年2期)2021-08-22 01:53:34
中學生數理化(高中版.高考數學)(2021年1期)2021-03-19 08:28:38
現代出版(2020年3期)2020-06-20 07:10:34
中華詩詞(2019年7期)2019-11-25 01:43:04
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
