續斷鑒別及川續斷皂苷VI的含量測定
2021-12-24 03:07:26艾光麗艾青青
亞太傳統醫藥 2021年12期
關鍵詞:方法
艾光麗,艾青青,高 鵬
(成都市食品藥品檢驗研究院/國家藥品監督管理局中藥材質量監測評價重點實驗室,四川 成都 610045)
續斷為川續斷科植物川續斷DipsacusasperWall.ex Henry的干燥根,具有補肝腎、強筋骨、續折傷、止崩漏的功效[1]。現代研究表明,續斷主要含有三萜皂苷、環烯醚萜苷、生物堿等成分[2-4],其中三萜皂苷類成分川續斷皂苷Ⅵ為主要活性成分,具有促進骨髓間質干細胞增殖和向成骨細胞分化以及促進骨傷愈合、抗骨質疏松的作用[5-7]。續斷主產于湖北、湖南、重慶、四川、貴州等省市[2-3]。《中國藥典》2020版第一部“續斷鑒別及含量測定”項下方法較為復雜,故本研究對其薄層鑒別及含量測定進行了改進,報道如下。
1 儀器與試藥
儀器:Waters2695-2998高效液相色譜儀(美國Waters公司);Agilent 1200高效液相色譜儀(德國安捷倫科技有限公司);Thermo UltiMate 3000高效液相色譜儀(美國賽默飛世爾科技有限公司);CAMAG自動點樣器;ME204E電子天平(瑞士梅特勒-托利多公司);SK250H超聲波清洗器;默克(Merck)HPTLC高效薄層層析板等。
對照品:川續斷皂苷VI(111685-201908,含量為94.3%);續斷對照藥材(121033-201812);均由中國食品藥品檢定研究院提供。
樣品:續斷樣品32批,其中四川23批、貴州2批、云南7批。
2 方法與結果
2.1 薄層色譜鑒別
2.1.1 供試品溶液的制備 取續斷粉末0.1 g,加20 mL甲醇,超聲處理20 min,作為供試品溶液。
2.1.2 對照藥材溶液的制備 取續斷對照藥材0.1 g,加20 mL甲醇,超聲處理20 min,作為對照藥材溶液。
2.1.3 對照品溶液的制備 取川續斷皂苷VI對照品,加甲醇制成每 1 mL約含 0.2 mg的溶液,作為對照品溶液。
2.1.4 點樣量 吸取續斷對照藥材溶液、續斷供試品溶液、川續斷皂苷VI對照品溶液各 10 μL,分別點于同一高效硅膠G薄層板上。
2.1.5 展開系統的篩選 (1)藥典方法:參照2020年版《中國藥典》一部“續斷”項下鑒別(2)的方法,發現此方法顯色不明顯且處理方法較繁瑣,故對其進行了改進。薄層色譜,見圖1。
吳參謀沒有跑,他讓手下弟兄迅速搶占有利地形,阻擊四周云集的鬼子,他深知自己擋不了鬼子多久,但只要多擋一分鐘,孔老一他們就多一分活著逃脫的希望。

注:1.川續斷皂苷VI;2.續斷對照藥材;3.CD-1;4.CD-2;5.CD-3;6.CD-4;7.CD-5;8.CD-6;9.CD-7;10.CD-8;11.CD-9;12.CD-10。圖1 藥典方法薄層色譜
(2)選定方法:將續斷對照藥材溶液、續斷供試品溶液、川續斷皂苷VI對照品溶液分別點于同一高效硅膠G薄層板上,以二氯甲烷-甲醇-水(14∶7∶2)的下層溶液為展開劑,展開,取出,晾干,噴以10%硫酸乙醇溶液,在105 ℃下加熱至斑點顯色清晰,分別置日光和紫外光燈(365 nm)下檢視。發現此方法下供試品與對照藥材相對應的斑點更多,顏色更清晰,分離效果更好,操作更簡便,且Rf值適中,故認為運用此方法分離續斷中的有效成分較為適宜。薄層色譜,見圖2、圖3。

注:1.川續斷皂苷VI;2.續斷對照藥材;3.CD-1;4.CD-2;5.CD-3;6.CD-4;7.CD-5;8.CD-6;9.CD-7;10.CD-8;11.CD-9;12.CD-10。圖2 選定方法薄層色譜日光圖
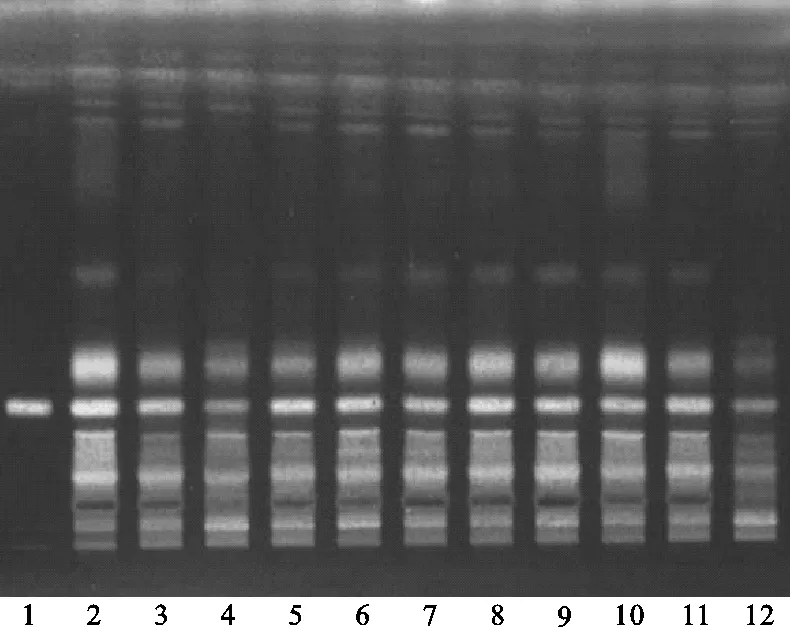
注:1.川續斷皂苷VI;2.續斷對照藥材;3.CD-1;4.CD-2;5.CD-3;6.CD-4;7.CD-5;8.CD-6;9.CD-7;10.CD-8;11.CD-9;12.CD-10。圖3 選定方法薄層色譜紫外圖
(3)其他薄層色譜展開條件篩選:方法①:將續斷對照藥材溶液、續斷供試品溶液、川續斷皂苷VI對照品溶液分別點于同一高效硅膠G薄層板上,以二氯甲烷-甲醇-水(14∶6∶2)的下層溶液為展開劑,此方法薄層色譜中供試品與對照藥材相對應的斑點多,顏色清晰,分離效果好,但川續斷皂苷VI的Rf值較低,故該展開系統不適合用于續斷定性鑒別。薄層色譜,見圖4、圖5。

注:1.川續斷皂苷VI;2.續斷對照藥材;3.CD-1;4.CD-2;5.CD-3;6.CD-4;7.CD-5;8.CD-6;9.CD-7;10.CD-8;11.CD-9;12.CD-10。圖4 方法①薄層色譜日光圖
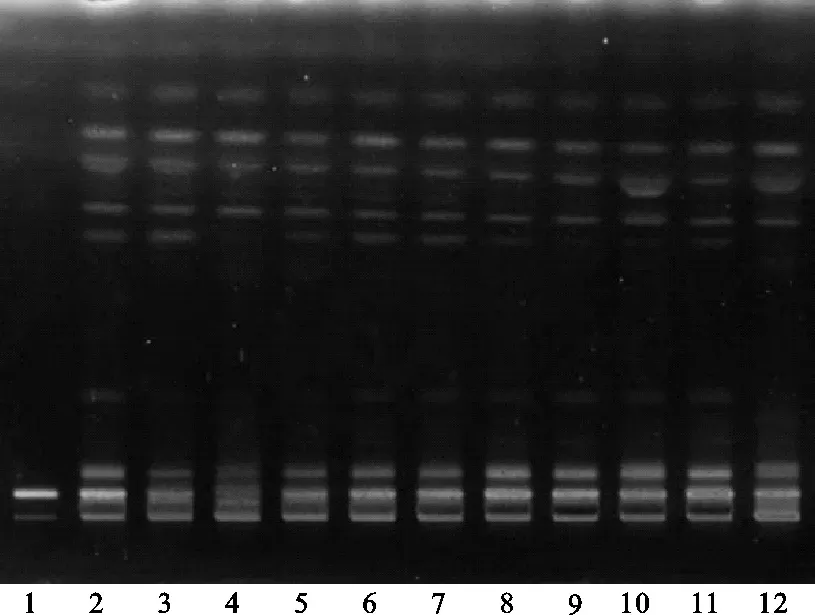
注:1.川續斷皂苷VI;2.續斷對照藥材;3.CD-1;4.CD-2;5.CD-3;6.CD-4;7.CD-5;8.CD-6;9.CD-7;10.CD-8;11.CD-9;12.CD-10。圖5 方法①薄層色譜紫外圖
方法②:將續斷對照藥材溶液、續斷供試品溶液、川續斷皂苷VI對照品溶液分別點于同一高效硅膠G薄層板上,以二氯甲烷-甲醇-水(13∶6∶2)[8]的下層溶液為展開劑,此方法薄層色譜中供試品與對照藥材相對應的斑點多,顏色清晰,分離效果好,但川續斷皂苷VI的Rf值較低,故該展開系統不適合用于續斷定性鑒別。薄層色譜,見圖6、圖7。

注:1.川續斷皂苷VI;2.續斷對照藥材;3.CD-1;4.CD-2;5.CD-3;6.CD-4;7.CD-5;8.CD-6;9.CD-7;10.CD-8;11.CD-9;12.CD-10。圖6 方法②薄層色譜日光圖
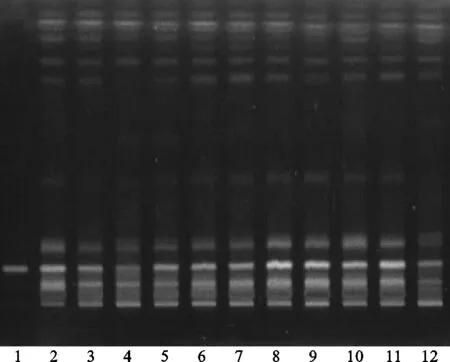
注:1.川續斷皂苷VI;2.續斷對照藥材;3.CD-1;4.CD-2;5.CD-3;6.CD-4;7.CD-5;8.CD-6;9.CD-7;10.CD-8;11.CD-9;12.CD-10。圖7 方法②薄層色譜紫外圖
方法③:將續斷對照藥材溶液、續斷供試品溶液、川續斷皂苷VI對照品溶液分別點于同一高效硅膠G薄層板上,正丁醇-乙酸-水(4∶1∶5)[9]的上層溶液為展開劑,此方法薄層色譜中供試品與對照藥材相對應的斑點顏色清晰,Rf值適中,但川續斷皂苷VI的分離效果差,故該展開系統不適合用于續斷定性鑒別。薄層色譜,見圖8、圖9。
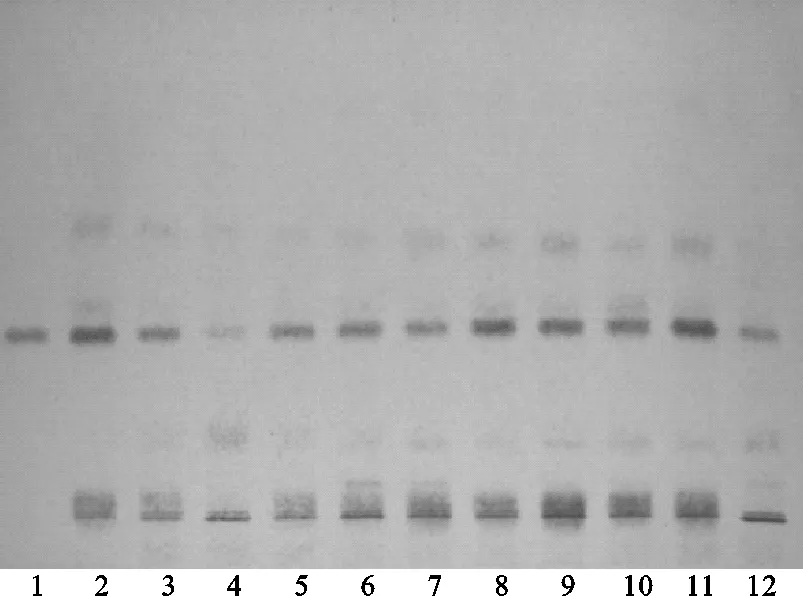
注:1.川續斷皂苷VI;2.續斷對照藥材;4.CD-1;4.CD-2;5.CD-3;6.CD-4;7.CD-5;8.CD-6;9.CD-7;10.CD-8;11.CD-9;12.CD-10。圖8 方法③薄層色譜日光圖
注:1.川續斷皂苷VI;2.續斷對照藥材;3.CD-1;4.CD-2;5.CD-3;6.CD-4;7.CD-5;8.CD-6;9.CD-7;10.CD-8;11.CD-9;12.CD-10。圖9 方法③薄層色譜紫外圖
方法④:將續斷對照藥材溶液、續斷供試品溶液、川續斷皂苷VI對照品溶液分別點于同一高效硅膠G薄層板上,正丁醇-冰醋酸-水(4∶1∶5)[10]的上層溶液為展開劑,此方法薄層色譜中供試品與對照藥材相對應的斑點顏色清晰,Rf值適中,但川續斷皂苷VI的分離效果差,故該展開系統不適合用于續斷定性鑒別。薄層色譜,見圖10、圖11。
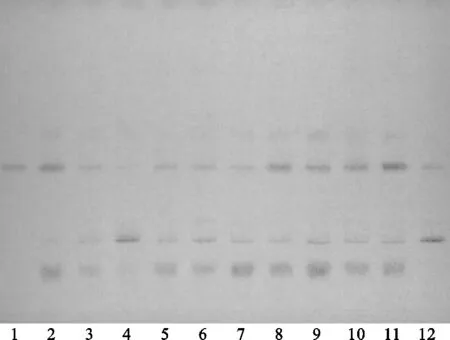
注:1.川續斷皂苷VI;2.續斷對照藥材;4.CD-1;4.CD-2;5.CD-3;6.CD-4;7.CD-5;8.CD-6;9.CD-7;10.CD-8;11.CD-9;12.CD-10。圖10 方法④薄層色譜日光圖

注:1.川續斷皂苷VI;2.續斷對照藥材;3.CD-1;4.CD-2;5.CD-3;6.CD-4;7.CD-5;8.CD-6;9.CD-7;10.CD-8;11.CD-9;12.CD-10。圖11 方法④薄層色譜紫外圖
2.2 川續斷皂苷VI的含量測定
2.2.1 色譜條件 以C18(250 mm×4.6 mm,5 μm)為色譜柱;以乙腈∶水(30∶70)為流動相;體積流量為1 mL/min;檢測波長212 nm,柱溫35 ℃,進樣量 10 μL。在上述色譜條件下,川續斷皂苷VI與其他色譜峰分離較好,分離度>1.5,且空白溶劑無干擾。見圖12。

注:A.對照品;B.樣品;C.空白溶劑;1.熊果酸。圖12 川續斷皂苷VI專屬性色譜圖
2.2.2 對照品溶液的制備 取川續斷皂苷VI對照品適量,精密稱定,加甲醇制成每1 mL含0.2 mg的對照品溶液。
2.2.3 供試品溶液的制備 取本品細粉0.5 g,置具塞錐形瓶中,精密加入甲醇100 mL,密塞,稱定重量,超聲處理(功率250 W,頻率40 kHz)20 min,放冷,再稱定重量,用甲醇補足減失的重量,搖勻,濾過,取續濾液,即得。
2.2.4 線性關系與標準曲線 精密稱取川續斷皂苷VI對照品7.915 mg,置10 mL容量瓶中,加甲醇使溶解并稀釋至刻度,作為對照品溶液。精密吸取上述溶液各0.1、0.2、0.3、0.4、0.5、0.6 mL,分別置10 mL容量瓶中,加甲醇稀釋至刻度。測定結果顯示川續斷皂苷VI進樣濃度在7.463 8~44.783 1 μg/mL范圍內與峰面積呈良好的線性關系;回歸方程:y=3446.113 5x-4 860.733 3;相關系數R2=0.999 9。
2.2.5 精密度試驗 精密吸取川續斷皂苷VI對照品溶液10 μL(0.182 2 mg·mL-1),注入液相色譜儀中,連續進樣6次,測得其峰面積并計算川續斷皂苷VI峰面積的RSD<3.0%,結果表明儀器的精密度良好。
2.2.6 穩定性試驗 精密吸取同一供試品溶液10 μL(批號CD-3),分別在0、6、10、12、14、16、20、24 h依次進樣,測定其峰面積并計算,川續斷皂苷VI峰面積的RSD<3.0%,結果表明供試品溶液在24 h 內穩定性良好。
2.2.7 重復性試驗 精密稱取同一批樣品6份(批號CD-3),按“2.2.3”項下方法制備供試品溶液,進樣10 μL依法測定峰面積并計算,川續斷皂苷VI含量的RSD為<3.0%,表明該方法重復性較好。
2.2.8 加樣回收率試驗 取已知含量的供試品(CD-2)9份,每份0.25 g,精密稱定,分成3組,每組3份,3組分別加入川續斷皂苷VI對照品溶液(濃度為4.640 7 mg/mL)0.5 mL,1 mL,1.5 mL。按擬定的方法制成供試品溶液,計算9份供試品的加樣回收率。結果顯示,川續斷皂苷VI的加樣回收率為101.37%~102.68%,RSD為0.45%,<3.0%,表明該方法準確度良好。見表1。

表1 川續斷皂苷VI加樣回收率試驗結果
2.2.9 樣品含量測定 取收集的32批續斷樣品,按選定方法測定川續斷皂苷VI的含量。見表2。

表2 32批續斷樣品含量測定結果
3 討論
續斷有效成分主要是皂苷或苷類成分,易溶于甲醇,故選擇甲醇作為提取溶劑。三氯甲烷、二氯甲烷對皂苷類成分分離效果較好,故選擇毒性相對較小的二氯甲烷作為展開劑的主要試劑。
取川續斷皂苷VI對照品溶液,在波長 200~400 nm 范圍內進行紫外光譜掃描,并記錄光譜圖。結果顯示其在近紫外端下具有較強吸收,參照2020年版《中國藥典》一部[1]“續斷含量”項下,故本研究選用 212 nm 作為檢測波長。
依據相關文獻方法[1,11-13],本實驗比較了乙腈-水(30∶70)、甲醇-0.1%鹽酸溶液(68∶32)、乙腈-0.05%甲酸溶液(30∶70)、乙腈-0.1%磷酸溶液(30∶70)四種流動相,結果以乙腈-水(30∶70)為流動相,基線平穩,色譜峰峰形對稱,操作簡便。
本實驗對提取溶劑(50%甲醇、70%甲醇、甲醇、乙醇)、超聲時間(20、30、40 min)、回流(30、40、60 min)等進行研究,在考察了分離度、色譜峰形以及過濾速度后,最終確定了甲醇超聲20 min作為供試品溶液的制備方法。
本實驗所建立的續斷薄層鑒別方法及川續斷皂苷VI的含量測定方法,方法簡便,專屬性強,耐用性好,能夠為續斷的質量控制和進一步開發利用提供實驗依據。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
意林原創版(2016年10期)2016-11-25 10:28:30
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
