自噬參與糖尿病血管病變的研究進展
2021-12-13 14:47:46魏海軍張鋮楊舒涵鄭楊申嘉陵劉勇何延政孫曉磊
中國比較醫學雜志 2021年11期
魏海軍張 鋮楊舒涵鄭 楊申嘉陵劉 勇何延政孫曉磊*
(1.西南醫科大學附屬醫院血管外科,四川 瀘州 646000;2.西南醫科大學心血管研究所,四川 瀘州 646000;3.四川省衛生健康委員會,成都 610041)
自噬(autophagy)是真核生物特有的一種分解代謝過程,在各種細胞內或細胞外刺激條件下,細胞質中受損的蛋白質、細胞器被雙層囊泡膜結構包裹后,與溶酶體融合,使得囊泡內容物降解以供細胞再利用。近年來,關于糖尿病血管損傷的病理生理機制有較多研究,其中高糖環境是導致糖尿病血管損傷的關鍵因素之一。而自噬作為一種機體的防御機制,在糖尿病血管損傷中發揮的作用不可小視。
1 自噬
1.1 自噬概述及其相關分子
自噬的過程包括:分隔膜、吞噬泡的形成,溶酶體與吞噬泡的融合,內容物的降解與釋放等過程。自噬的生理功能主要體現在以下幾個方面:(1)胞內蛋白可通過自噬被降解,實現細胞器的更新和轉化,從而維持細胞內環境的穩態[1];(2)由自噬產生的降解產物可被機體循環再利用以提供能量[2];(3)自噬作為一種防御機制,能在外界不良刺激和應激條件下產生適應性反應,保護受損的組織或細胞。但是,當自噬被過度激活時,又可作為一種程序性死亡調控程序,調控細胞的死亡[2]。自噬主要以三種不同的形式存在:巨自噬,微自噬和伴侶介導的自噬[3-4],所有這些都促進溶酶體中細胞溶質的蛋白水解降解。巨自噬和微觀自噬都能夠通過選擇性和非選擇性機制吞噬大型結構分子。在伴侶蛋白介導的自噬(CMA)中,靶向蛋白與伴侶蛋白(例如Hsc-70)形成復合物,穿過溶酶體膜,該蛋白被溶酶體膜受體溶酶體相關膜蛋白2A(LAMP-2A)識別,從而導致蛋白的變性和退化。
自噬相關基因(autophagy related gene,Atg)負責調控自噬的過程,它的基因片段在進化上相對保守。自噬的調控機制十分復雜,它包含Atg及其所編碼的蛋白產物參與自噬的誘導,分隔膜的形成,自噬體的產生、成熟等過程。ULK1復合物(ULK1、FIP200、Atg13)是連接上游刺激信號與下游自噬體形成的橋梁,能接收上游各種信號刺激并做出應答[5],從而誘導自噬產生。而PI3K復合物(Atg6/Beclin-1、Atg14、Vps34/PI3KC3、Vps15)可介導分隔膜的產生[6]。另外,Atg12的結合過程與微管相關蛋白1輕鏈3(microtubule associated protein 1 light chain 3,LC3)的修飾過程,這兩個泛素化過程在自噬體的延伸階段發揮著重要作用[7]。LC3是由哺乳動物細胞酵母菌Atg8編碼產生的一種自噬相關蛋白,被認為是一種自噬標志分子。前體LC3合成之后,被水解切割掉特異片段后,形成LC3I。LC3I又在Atg7和Atg12-Atg5-Atg16 L的作用下,轉變為LC3II,始終結合于自噬體膜上,所以LC3II的含量也可以反映自噬體的數量。Beclin-1是由哺乳動物細胞酵母菌Atg6編碼產生的一種自噬相關蛋白[8],可介導自噬蛋白定位于吞噬泡,繼而調控自噬體的形成與成熟,其表達量的多少亦可反應自噬活性的強弱。
1.2 糖尿病狀態下自噬調控的分子機制
目前的研究中,糖尿病通過以下通路影響細胞的自噬:氧化應激反應(oxidative stress)、內質網應激(ER stress)、mTOR依賴性信號通路、AMPK途徑等。
2型糖尿病的特征是高血糖和高胰島素血癥,通常伴有糖原和脂質的細胞內蓄積。糖基化和脂肪酸中間體的積累破壞了線粒體和過氧化物酶體的穩定性,導致產生活性氧[9]。并且營養物質的過量也會促進未折疊蛋白和潛在毒性脂質池的形成,從而引起內質網應激[10-11]。自噬通過去除部分去極化的線粒體,將ROS水平維持在一定的水平,另一方面,ROS可以通過抑制mTOR、增加Beclin-1的表達、LC3-I向LC3-II的轉化激活自噬[12-15]。此外,ROS可以作為激活JNK-1的信號分子[16],大量的ROS導致線粒體通透性轉變孔的開放使線粒體膜電位的破壞,從而導致PINK1/parkin介導的自噬發生[17-19],ROS還可以通過轉錄水平(在細胞核中)和翻譯水平(在細胞質中)調節來誘導自噬過程[20],ROS升高分別激活轉錄因子HIF-1α、p53、FOXO3、Nrf2,促進BNIP3、NIX、TIGAR、LC3和p62的轉錄[21]。
內質網應激通過UPR途徑和內質網Ca2+轉運來誘導自噬[22]。UPR的三個途徑:(a)PERK通路——它的激活導致轉錄因子4(transcription factor 4,ATF4)的翻譯[23],(b)IRE1α通路——激活c-Jun-N-terminal激酶(JNK途徑[24]和通過移碼突變的方式修飾x-box結合蛋白1(x-box binding protein 1,XBP1)的mRNA[25],(c)ATF6通路——調節其他UPR成員包括XBP1[26]。PERK通路通過ATF4誘導ATG5、ATG7、ATG10等自噬相關基因的轉錄從而影響自噬[27]。PERK通路也促進了聚谷氨酰胺72(polyglutamine 72,PolyQ72)誘導LC3-I向LC3-II的轉化過程[28]。在內質網應激的早期階段,激活的IRE1α通過JUK途徑磷酸化參與誘導自噬過程的Bcl-2,從而導致Bcl-2從Beclin-1分離出來[29]。此外,XBP-1—IRE1α途徑的另一個參與者,也會促進Beclin-1轉錄。ATF6通過誘導XBP-1的轉錄間接參與自噬過程[22]。ER應激下,過度的Ca2+進入細胞質中通過3個不同的機制誘導自噬:(a)刺激依賴CamKK/AMPK途徑導致mTOR抑制[30],(b)激活的壞死相關蛋白激酶(death-associated protein kinase,DAPK)通過參與磷酸化Beclin-1[31],(c)激活PKC途徑從而導致Bcl2-Beclin-1復合體分離。
高血糖可激活mTOR信號通路減輕細胞自噬。mTOR的機制靶標是經典的營養途徑,通過與兩種不同的蛋白復合物mTOR復合物1(mTORC1)和mTOR復合物2(mTORC2)結合來調節自噬活性。其中,mTORC1通過直接磷酸化抑制Ulk1復合物的活性,從而抑制自噬相關蛋白LC3B和Beclin-1的表達[32-33]。mTORC1在高血糖情況下被很大程度的激活[34]。通過特異性地敲低相關基因或者使用雷帕霉素(mTOR的抑制劑)抑制mTORC1的表達,可以促進自噬的恢復可以減輕糖尿病對細胞的產生的不利影響[35]。
AMP激活的蛋白激酶(AMPK)是一種營養敏感的激酶,在葡萄糖不足的情況下,AMPK通過Ser317和Ser777的磷酸化直接激活ULK1,從而促進自噬。相反,高葡萄糖下的mTOR或AKT活性增強導致Ser757處的ULK1磷酸化,抑制ULK1活化,從而破壞ULK1和AMPK之間的相互作用,并減少AMPK下游信號通路的自噬誘導作用[32]。AMPK的激活還可以在Beclin-1與BCL-2分離后刺激JNK1形成VPS34蛋白復合物17,并激活自噬[36]。其他研究也報道,AMPK調節FoxO轉錄因子的表達,從而誘導自噬相關基因的表達[37]。
2 自噬參與糖尿病相關血管病變
2.1 微血管病變
2.1.1 自噬在糖尿病視網膜病變中的作用
糖尿病視網膜病變(diabetic retinopathy,DR)是一種糖尿病微血管并發癥,占致失明原因的5%,其組織學特征包括血-視網膜屏障破裂、新生血管形成、毛細血管無灌注、周細胞脫落、內皮細胞減少和纖維血管增生異常。研究表明,毛細血管閉塞和毛細血管通透性的增加將破壞血-視網膜屏障,是產生血管滲漏的主要原因,而血管內皮生長因子(vascular endothelial growth factor,VEGF)既可促進血管滲漏,又可導致新生血管生成,在DR中扮演了重要的角色。近幾年的研究中表明,自噬在糖尿病視網膜病變的新生血管的形成以及血管滲漏中起著重要的作用,而自噬可以調節VEGF的分泌從而減輕相應的病變。
高糖環境可以使細胞中的ROS水平增高,導致炎癥反應,從而使釋放TNF-α、IL-1β、IL-17A、IL-8和IL-6,在這些炎癥介質水平的升高的同時,VEGF連同其可溶性受體sIL-2R的表達也相應增加,VEGF的升高促進新生血管的形成使DR進一步加重[38]。Müller細胞是糖尿病視網膜病變中分泌VEGF較多的細胞之一,研究發現高糖水平可以促進Müller細胞中的自噬標記物升高,但是p62/SQTSM1與內質網應激水平也持續升高,其結果導致溶酶體功能失調使得自噬功能障礙而導致細胞的凋亡,VEGF的釋放增加[39];通過轉染siRNA抑制p62/SQTSM1的表達以及mTOR的抑制劑Rapamycin,激活cathepsin L的活性使溶酶體水解活性增強,導致自噬體降解,使細胞的自噬功能恢復從而抑制VEGF的分泌。與此類似的研究也說明[40],高糖導致的p62/SQTSM1的表達升高,可以被三七皂苷(notoginsenoside R1,NGR1)通過PINK1依賴的線粒體自噬途徑抑制,降低Müller細胞中的ROS水平以及炎癥反應,從而降低VEGF的表達;NGR1上調了HG誘導的rMC-1細胞和db/db小鼠視網膜中PINK1和Parkin的水平,增加了LC3II/LC3I的比例,并下調了p62/SQSTM1的水平,從而減少細胞中的ROS水平以及炎癥反應,降低VEGF的表達。
高血糖除了可以直接影響p62/SQTSM1的表達以外,還可以損傷血-視屏障導致低密度脂蛋白(LDL)從血管中滲出。經過氧化以及糖基化修飾后的低密度脂蛋白(HOG-LDL)在早期的DR中有很強的驅動作用。然而在這個過程中,誘發的自噬會導致細胞出現死亡以及VEGF的分泌,導致新生血管形成從而加重糖尿病視網膜病變。研究發現Müller細胞經過HOG-LDL刺激后使AMPK激活導致自噬相關蛋白ATG-5、Beclin-1、LC3II/LC3I蛋白水平明顯升高,與此同時凋亡相關蛋白caspase-3的表達也進一步升高說明自噬的激活會導致細胞凋亡[41]。他們的研究中使用芐基四羥基喹啉(benzyltetrahydroxyquinoline,berberine)可以抑制AMPK的激活減輕細胞中HOG-LDL導致的自噬性細胞死亡、炎癥反應以及血管新生等。近幾年的一些研究通過高糖培養視網膜內皮細胞(retinal endothelial cells,RECs)來建立糖尿病視網膜的血管新生病變的體外模型來進一步探尋DR的內在機制。Mao等[42]研究提示去乙酰化酶3(Sirtuin 3,Sir3)可以抑制HRECs中遷移相關因子MMP-2、MMP-9、血管新生因子VEGF、HIF-1a、和IGF-1的表達,而且研究中發現過表達Sir3可以促進LC3I以及LC3II的表達以促進細胞自噬,并且細胞自噬后期可以降低VEGF、HIF-1α的表達,故Sir3可能通過自噬來抑制HRECs的血管新生。Mao等[43]進一步研究發現在體內實驗中和體外實驗中發現miR-204-5p可以促進糖尿病大鼠視網膜的血管新生,其中LC3II以及LC3I/LC3II的表達降低,說明通過降低miR-204-5p的表達可以促進LC3II的表達從而減低VEGF的分泌。
綜上所述,自噬在糖尿病視網膜病變中在多個細胞中起著調節作用,目前大部分的研究肯定了自噬在DR中的保護作用,但是在某些情況,過度自噬也將會導致DR進一步惡化[41],這兩種作用的相互聯系尚需進一步研究闡明。
2.1.2 自噬在糖尿病腎病中的作用
糖尿病腎病(diabetic nephropathy,DN)作為糖尿病最嚴重的并發癥之一,目前已成為繼腎小球腎炎后終末期腎病的第2位原因,DN的一個重要臨床特征是尿蛋白。在蛋白尿的糖尿病患者中發現腎小球濾過屏障的結構改變,包括腎小球內皮細胞損傷[44-45]。近來有越來越多的證據表明,腎小球血管周圍的足細胞自噬水平對糖尿病腎病發展有重要的作用。在不同類型的細胞中,自噬發揮的作用也有所不同:
(1)足細胞 足細胞損傷是DN中關鍵因子,可以作為DN演變的臨床預測指標。在高糖的反復刺激下,維持細胞內穩態對于足細胞的命運極為重要[46]。近幾年的研究中發現,自噬可以維持足細胞的穩態,從而改善糖尿病腎病。研究發現,相當一部分腎保護藥物是通過mTOR/AMPK途徑促進自噬相關蛋白如LC3II、Beclin-1等的表達,增強自噬形成,從而對足細胞產生保護作用[47-53]。Liu等[54]研究發現,在高血糖下,足細胞中β-arrestin-1和β-arrestin-2的表達水平增高,而且β-arrestin-1和β-arrestin-2可以通過抑制ATG12-ATG5的結合來抑制足細胞自噬,導致腎的損傷,這可能成為治療糖尿病腎病新的靶點。
既往的研究顯示,在糖尿病腎病的動物模型中發現了上皮-間充質轉換(epithelial-mesenchymal transition,EMT)的病理現象。EMT是一個動態的發展過程,其特征在于上皮細胞獲得間充質細胞(如成肌纖維細胞)的特征,這會導致上皮細胞標記物(如E-鈣黏著蛋白)表達降低,間充質標記物(如α-SMA和纖連蛋白)的表達增強,從而破壞腎小管基底膜以及增強足細胞的遷移[55]。Jin等[56]發現高糖培養下,足細胞中STIM1激活TRPC6和Orai1通道,使鈣離子通道打開,導致p62表達增高和LC3II/LC3I降低從而抑制自噬,最終加速了EMT的發生。Jin等[57]在進一步研究后發現,HMGB1通過激活AKT/mTOR通路抑制LC3II的表達和促進p62的升高,從而導致足細胞的EMT的增強。這些研究說明,自噬抑制足細胞的EMT,從而抑制糖尿病腎病的進展。
(2)腎小球內皮細胞 Lenoir等[58]發現內皮細胞自噬與糖尿病腎病之間相關的直接證據。他們研究顯示,內皮細胞特異性自噬缺陷小鼠會出現嚴重的腎小球損傷,表明內皮細胞自噬對糖尿病所引起的應激起著重要的保護作用;并且內皮細胞特異性自噬缺陷小鼠模型可提供足細胞和近端腎小管細胞以外的其他腎細胞中糖尿病腎病自噬的重要信息。晚期糖基化終末產物(AGEs)和AGEs受體(RAGE)被證明在糖尿病性腎病中起重要作用。Hou等[59]研究表明丹酚酸A(salvianolic acid A,SalA)通過AGE-RAGE-Nox4軸減弱了AGEs誘導的ROS,從而促進Atg5、Atg7、Atg12、LC3II/LC3I的表達以及抑制p62的表達,從而恢復高糖導致的內皮細胞自噬紊亂,抑制DN的進程。Lim等[60]的研究表明,西那卡塞(Cinacalcet)通過CaMKKβ-LKB1-AMPK軸促進腎小球內皮細胞中LC3II/LC3I、Beclin-1的表達,從而減輕高糖對腎小球的損傷。這些研究證明,腎小球內皮細胞中自噬的激活對腎臟功能起著保護作用。
2.2 外周血管病變
2.2.1 自噬在糖尿病動脈粥樣硬化性病變中的作用
動脈粥樣硬化是導致心腦血管疾病以及下肢動脈硬化的主要原因,它是以脂質蓄積和炎癥細胞浸潤為特征的慢性炎癥性過程。在糖尿病導致的動脈硬化疾病中,自噬起著調節作用。Tian等[61]的研究發現,高糖激活mTOR信號通路抑制LC3II/LC3I以及Beclin-1的表達以及增加SQSTM1/p62堆積抑制自噬,導致內質網應激相關蛋白如p-JNK、CHOP和Caspase-12表達升高,誘導凋亡,從而促進鏈脲菌素誘導的糖尿病ApoE-/-小鼠動脈粥樣硬化。Tian等[62]的研究發現,糖尿病狀態下,糖化高密度脂蛋白引起巨噬細胞的凋亡并伴隨內質網應激活化—包括核轉位激活轉錄因子6、蛋白激酶樣ER激酶(PERK)的磷酸化和真核生物翻譯起始因子2以及C/EPB同源蛋白的上調,通過加入自噬誘導劑雷帕霉素(Rapamycin,RAPA)可以抑制上述過程,從而抑制巨噬細胞的凋亡;而加入自噬的抑制劑3-MA或抑制Beclin-1表達可以促進這一過程,導致巨噬細胞的凋亡,從而促進糖尿病動脈粥樣硬化性疾病的進展。Lazaro等[63]的實驗也證明核樣因子2(nuclear factor(erythroid-derived 2)-like 2)可以通過促進自噬相關蛋白Beclin-1、LC3II/LC3I的表達升高,SQSTM1表達的降低來抑制血管壁的炎癥相關水平,使糖尿病導致的動脈粥樣硬化向好的方向發展。
動脈粥樣硬化的始發事件是血管內皮細胞的損傷[8]。高血糖可導致內皮細胞內的活性氧水平上升,從而導致炎癥,內皮細胞的損傷最終導致細胞凋亡。Xie等[64]的研究中發現,AGEs可以使血管內皮細胞中的活性氧水平升高,作為一種防御機制,機體內的自噬水平相應的增加,而且自噬相關的抑制劑3-MA加重了AGEs對內皮細胞的損傷。Fetterman等[65]研究發現在內皮細胞中,自噬可以激活一氧化氮合酶(eNOS);通過從糖尿病病人以及正常人中收集內皮細胞分析發現,相比于正常人來講,糖尿病病人內皮細胞的p62水平明顯升高,標志著自噬通量降低。雖然糖尿病組與正常組的LC3以及Beclin-1水平相似,但是在糖尿病組中的自噬終末期的標志物溶酶體蛋白Lamp2a的水平明顯升高,通過抑制終末期的自噬,使自噬長期處于活躍水平導致eNOS激活,從而減輕內皮細胞的損傷。Bai等[66]的研究中提出高血糖導致動脈硬化的新機制,他們的研究指出內皮細胞中的小窩蛋白1(caveolin-1,CAV1)具有介導脂質大分子物質胞吞的作用,它有兩種類型的LC3相互作用區(LC3-interacting region,LIR):CAV1支架結構域(the CAV1 scaffolding domain,CSD)和膜內結構域(intramembrane domain,IMD)。CAV1可 以 與CAVIN1-LC3B結合并形成復合物。由于小窩相關蛋白1(caveolae associated protein 1,CAVIN1)沒有LC3結合片段,而CAV1具有超過2個LIR,所以CAV1作為CAVIN1和LC3B之間的中間連接體可以介導自噬抑制。當CAV1作為結構蛋白存在于膜小窩或小窩中以介導LDL轉胞作用時,IMD中的LIR被掩埋,CAV1僅對通過CSD的自噬發揮抑制作用。當CAV1存在于細胞質中時,CAV1通過IMD可能有機會與LC3B結合并介導CAV1的自噬降解。在正常的葡萄糖環境下,CAV1在細胞質中的自噬降解和利用CAV1形成膜小泡保持了一定的穩態,并且在內皮細胞膜中小泡的形成相對較少,因此內吞小窩對LDL的胞吞作用也維持在相對較低的水平。通過抑制AMPK-mTOR-PIK3C3途徑,高葡萄糖抑制了CAV1-CAVIN1-LC3B介導的CAV1自噬降解。結果,更多的CAV1積累在細胞質中,在細胞膜中形成更多的小孔并促進LDL跨內皮細胞的胞吞作用,從而促進動脈粥樣硬化脂質在內皮下的滯留,這個機制或許可為糖尿病導致動脈粥樣硬化提供新的治療方案。
與此同時,其他的一些研究也發現自噬在內皮功能保護中的不利作用。Cai等[67]的研究發現,與正常組比較,高糖組自噬相關蛋白Atg7、LC3II/LC3I的水平明顯升高,而且這個過程中凋亡相關蛋白Bax、BCL-2蛋白水平也明顯升高,說明高糖可能導致內皮細胞的自噬性死亡,而GLP-1高血糖素樣肽-1(glucagon-like peptide-1,GLP-1)通過GLP-1RERK1/2依賴性通路,恢復HDAC6活性,抑制高糖導致內皮細胞功能障礙和過度自噬性死亡,提示自噬在內皮細胞中有害的一面。除此之外,Qiu等[68]發現,高糖降低血管平滑肌細胞內硫化氫的水平,激活AMPK/mTOR通路導致LC3II/LC3I、Beclin-1的水平升高以及P62降低,而這個自噬增強的過程可以導致平滑肌細胞的功能紊亂,也提示了自噬在糖尿病動脈粥樣硬化過程中的不利作用。
2.2.2 自噬在糖尿病血栓形成以及瘤樣擴張狀態中的作用
大約65%的糖尿病患者死于血栓性事件,包括心臟病發作和中風[69]。其中大約40%~60%的糖尿病病人對抗血栓藥物阿司匹林耐受[70],進一步的研究發現糖尿病病人血液中血小板被顯著激活[71]。Lee等[71]研究發現,高糖可以導致血小板內的ROS水平升高,pp53的激活,線粒體功能紊亂,導致血小板的凋亡從而形成血栓;而激活線粒體自噬可以維持線粒體功能抑,制血小板的功能紊亂,從而抑制血栓的形成。Paul等[72]通過饑餓的方式抑制血小板的聚集,自噬相關的因子Atg3、Atg5、Atg7、Atg12、Atg16、Beclin-1、LC3II/I明顯上升,加入自噬抑制劑3-MA可以恢復血小板的聚集,進一步證實了自噬在血小板的聚集中的有利的作用。
主動脈中膜平滑肌細胞的減少導致中膜薄弱可以導致動脈瘤的形成,糖尿病狀態下,腹主動脈瘤與胸主動脈瘤的發病率明顯降低[73]。Zheng等[74]發現,腹主動脈瘤患者組織中自噬相關基因轉錄因子TG4b、Atg6/Beclin-1,Bnip3和Vps34等表達升高,伴隨整合素/CD44的激活和p38/MAPK信號通路的增強,同時,LC3蛋白表達水平且LC3II/I值也相應增高。另外,Giusti等[75]通過微陣列分析技術對腹主動脈瘤患者和正常人的外周血進行基因表達譜差異分析,發現自噬相關基因Atg5表達上調。因此,腹主動脈瘤患者中自噬水平的升高很可能與機體自身的防御抵抗機制密切相關。近來又有研究發現,RAPA可極大地延緩腹主動脈瘤的發病進程,但RAPA同時也是一種炎癥抑制劑,故不能完全確定RAPA是否通過激活自噬而減輕腹主動脈瘤的進展[76]。這些研究都證實自噬和高血糖可能參與腹主動脈瘤的病理過程,但目前尚沒有更多研究闡明自噬、糖尿病以及動脈瘤之間相互關系。
上述研究結果顯示,雖然自噬相關基因在糖尿病外周血管病變中上調,但自噬在糖尿病外周血管病變發病機制中的具體機制仍未闡明。體外實驗中,許多糖尿病外周血管病變相關因子可以激活自噬而發揮保護性作用,且此保護作用是通過減弱氧化應激和炎癥信號轉導而起效;體內實驗中,越來越多的證據表明自噬在動脈粥樣硬化中起保護作用,且可維持動脈粥樣硬化斑塊的穩定性。其中,RAPA減輕糖尿病外周血管病變的機制尚不清楚,但其為激活自噬治療糖尿病外周血管病變提供了新的方向。
3 結語與展望
作為機體的防御機制和蛋白質降解的重要途徑,自噬在糖尿病血管病變中的作用正在漸漸被探索、挖掘。本文綜述了自噬分別在糖尿病視網膜病變、糖尿病腎病、糖尿病外周血管病變、糖尿病心腦血管病變中的作用,并對自噬參與糖尿病血管病變的分子生物學機制進行了深入探討(圖1)。就目前的研究趨勢來看,通過使用自噬激活劑和抑制劑來調控自噬的水平已經成為了學者們的慣用思路,當然,過表達特定自噬相關基因或使其被敲除也是目前常用的研究方法。但是大多數研究都是從細胞水平出發探究自噬在相關病理狀態下的作用,也使得其后續的功能檢測方法受限,因而建議從整體水平更加全面地探究自噬在糖尿病血管病變中的作用。并且,自噬在某些糖尿病血管病變中的病理生理作用仍未闡述清楚,部分結論仍有爭議,所以未來仍需深入研究。糖尿病血管病變的病理生理學機制十分復雜,因此找尋其發病機制中的關鍵因子或重要的信號通路就顯得尤為重要,通過自噬篩選的相應靶點可以作為治療糖尿病血管病變的新途徑。雖然目前自噬在糖尿病血管病變中的發生機制仍未闡明,但相信隨著研究的不斷深入,靶向自噬的機制研究一定會為糖尿病血管病變的治療開創新局面。
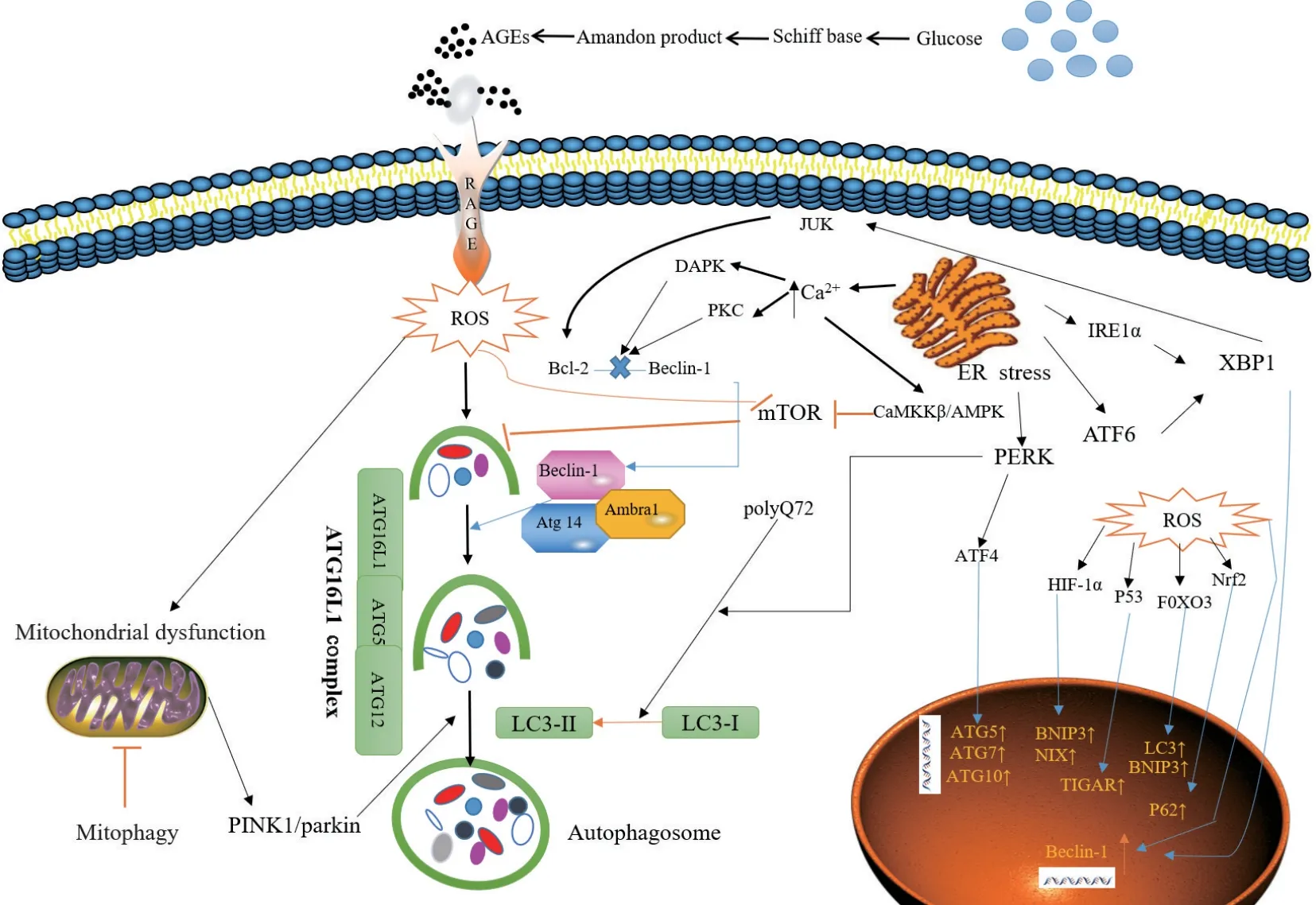
圖1 糖尿病狀態下自噬調節的信號通路Figure 1 Signal pathways regulating autophagy in diabetes
猜你喜歡
中老年保健(2022年5期)2022-08-24 02:35:42
中老年保健(2022年1期)2022-08-17 06:14:56
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
中老年保健(2021年5期)2021-08-24 07:07:20
中老年保健(2021年11期)2021-08-22 03:15:16
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
人大建設(2019年12期)2019-05-21 02:55:32
