固相萃取/氣質聯用法測定水中乙草胺的方法探討
2021-12-09 01:06:40鄧智友王建蓉周智勇蔡雨含
供水技術 2021年5期
鄧智友, 王建蓉, 周智勇, 胥 川, 蔡雨含, 馬 懿
(四川省城市供排水水質監測網綿陽監測站, 四川 綿陽 621000)
乙草胺(Acetochor)是一種選擇性芽前酰胺類除草劑,廣泛應用于花生、玉米等豆類植物,但由于乙草胺及其代謝產物醌亞胺具有致癌作用且容易殘留,已被美國國家環境保護局(USEPA)定為B-2類致癌物[1-2]。乙草胺會經過雨水、灌溉等方式進入水體從而造成水體污染,目前國內外農業以及水質檢測方面對乙草胺的檢測方法報道較少,因此有必要建立一種快速可靠檢測乙草胺的方法。
1 方法原理
水樣中乙草胺被Waters Oasis HLB固相萃取小柱富集吸附,依次用不同溶劑洗脫,洗脫液經濃縮后用氣相色譜毛細柱分離,然后進入質譜儀檢測。通過目標組分的質譜圖和保留時間與標準物質的質譜圖和保留時間作對照進行定性,目標組分定量離子的響應值大小與濃度成正比,以此進行水中有機化合物的定量。
2 實驗部分
2.1 實驗儀器與試劑
2.1.1 實驗儀器
氣相色譜-質譜(GC-MS)儀:帶自動進樣器的Agilent 7890A GC-5975C MS,配工作站;固相萃取儀Supelco VISIPREP DL;無油真空泵Lab Tech;抽濾瓶。
2.1.2 標準溶液
乙草胺Acetochlor(34256-82-1)標準溶液:100 μg/mL;色譜純有機溶劑:甲醇,乙酸乙酯,丙酮;硫酸:1+3硫酸溶液;亞硫酸鈉(2%),優級純。
2.1.3 實驗材料
移液管:5和10 mL;C18、Waters HLB固相萃取小柱;微量注射器:10 μL。
2.2 GC-MS檢測條件
2.2.1 氣相色譜條件
Agilent 190915-433毛細管柱(30 m×0.25 mm×0.25 μm);色譜柱溫度:100℃保持2 min,然后以10℃/min 升溫至200℃,再以30℃/min升溫至270℃,保持10 min;不分流進樣方式;進樣量:1.0 μL;進樣口溫度: 250℃;載氣:氦氣(純度≥99.999%);流速:1 mL/min。
2.2.2 質譜條件
色譜-質譜接口溫度:280℃;電離方式:EI;電子能量:70 eV;離子源溫度:230℃;四級桿溫度:150℃;溶劑延遲:3.6 min。
選擇SIM離子模式,在NIST標準譜庫中檢索到乙草胺的EI 質譜圖,其特征碎片離子主要為m/z 59,132,117,146,162,174,223,但是m/z 59 的碎片過小,對其干擾明顯,故不采用。根據除草劑中檢測乙草胺的相關文獻[3],最終選擇146為定量離子,確定146,162,223為定性離子。
2.3 水樣前處理和固相萃取小柱活化
采集自來水水樣時,先放水數分鐘以排除水管中存水,再用采樣瓶采集水樣。加入約2%亞硫酸鈉1 mL,蓋好瓶蓋后混勻,靜止數分鐘去除余氯待萃取。
固相萃取小柱活化條件:固相萃取柱預先用5 mL甲醇、5 mL純水過柱,注意不讓甲醇和水流干。
2.3.1 萃取吸附
取500 mL已去除余氯且混合均勻的水樣,用1+3硫酸溶液約1.5 mL調節pH值為1.5左右,混勻后用HLB固相萃取小柱進行萃取。控制真空泵壓力,萃取水樣以2 mL/min的流速通過活化后的HLB小柱,進行固相萃取。
2.3.2 洗脫與濃縮
結合其化學分子結構,乙草胺是一種極性化合物,在非極性的正己烷難溶。根據這一特點,先后采用極性比較好的不同溶劑組合乙酸乙酯+丙酮(1 ∶1)、丙酮+甲醇(1 ∶1)、丙酮、甲醇對其小柱進行洗脫;洗脫液濃縮至1 mL,待氣相色譜質譜儀檢測用。
2.4 定性分析
通過目標組分的質譜圖和保留時間與標準物質的質譜圖和保留時間作對照進行定性(保留時間隨柱長不同而略有變化),乙草胺保留時間在14 min,見圖1。目標組分定量離子的響應值大小與濃度成正比,以此確定水中乙草胺含量。
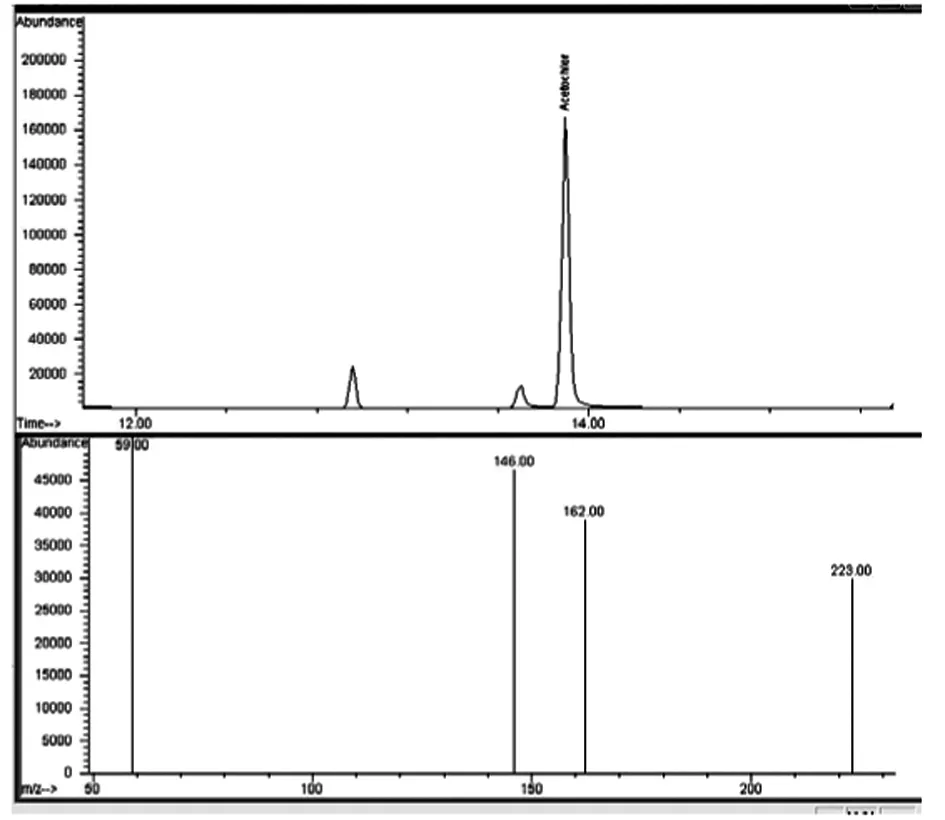
圖1 乙草胺標準的總離子色譜
2.5 標準曲線的繪制
用乙草胺標準物質配成不同濃度梯度的標準系列,在與待測樣品完全相同的操作條件下,分別在給定的色譜質譜條件下進行測定。
由于低濃度與高濃度的水平梯度跨度較大,繪制成一個標準曲線的線性效果較差,因此將標準物質分別按照低濃度和高濃度范圍配制成標準系列。以目標化合物峰面積為縱坐標、濃度為橫坐標繪制標準曲線,曲線線性良好,如表1所示。
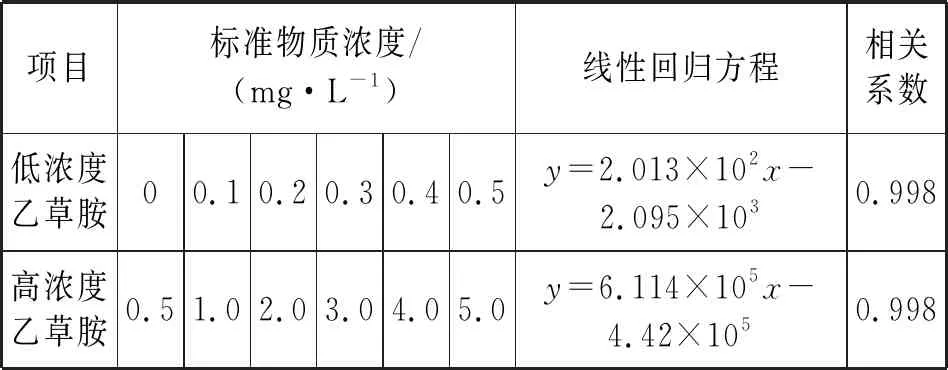
表1 標準系列和線性方程
2.6 萃取小柱與洗脫溶劑
用乙草胺標準物質加標濃度2 μg/mL至純水平行水樣中,在與待測樣品完全相同的操作條件下,分別用C18小柱與HLB小柱進行萃取,萃取結束后分別按照不同組合溶劑乙酸乙酯+丙酮(1 ∶1)、丙酮+甲醇(1 ∶1)、丙酮、甲醇對其小柱進行洗脫,分別在給定的色譜質譜條件下進行測定。
3 結果與討論
3.1 線性范圍、檢出限和測定下限
在試驗所采用的色譜和質譜條件下,乙草胺分離情況和響應較好,濃度在0~0.5 mg/L內線性良好,相關系數為0.998;0.5~5.0 mg/L內呈良好線性,相關系數為0.998。
測定7個平行加標濃度為0.10 μg/L的乙草胺水樣,測定結果分別為105,97,103,94,107,110和96 ng/L。計算得到標準偏差Si=6.1 ng/L,檢出限[3]MDL=19.2 ng/L,測定下限LOD=77 ng/L。
3.2 萃取小柱與洗脫溶劑的選擇
從表2可以看出,加標回收(加標2 μg/mL)實驗檢測結果受柱子的影響不大,因此實驗選擇HLB小柱。對萃取實驗影響較大的因素為溶劑的選擇,乙酸乙酯+丙酮(1 ∶1)和丙酮洗脫的回收率相差不大且效果明顯;丙酮+甲醇(1 ∶1)和甲醇單獨對其小柱進行洗脫,洗脫效果相當且較差,所以實驗選擇洗脫方式為乙酸乙酯+丙酮(1 ∶1)或者丙酮洗脫。
3.3 精密度與準確性評估
分別取500 mL自來水水樣各7份,進行低濃度2.0 μg/L和高濃度8.0 μg/L水平的加標回收試驗,結果見表3。乙草胺加標回收率在90.5%~112.5%,相對標準偏差RSD為4.5%~4.8%。
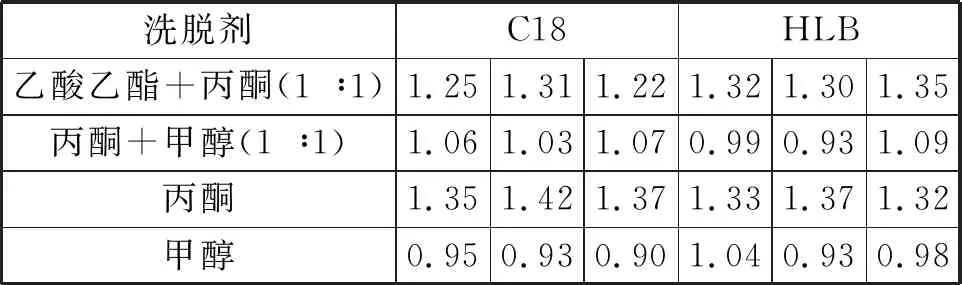
表2 不同柱子萃取與溶劑洗脫的回收率
4 結論
采用固相萃取氣相色譜-質譜法測定水樣中的乙草胺,獲得對稱平滑正態分布的色譜峰,精密度較好,相對標準偏差在4.5%~4.8%,加標回收率在90.5%~112.5%,該方法準確、穩定、簡單可靠。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
海峽科技與產業(2016年3期)2016-05-17 04:32:12
