抑制劑在銅表面吸附機理的實驗與理論研究進展
2021-12-01 01:15:32李偉檀柏梅馬騰達紀金伯閆妹張詩浩王亞珍
電鍍與涂飾 2021年21期
李偉 ,檀柏梅 , ,馬騰達 ,紀金伯 ,閆妹 ,張詩浩 ,王亞珍
(1.河北工業大學電子信息工程學院,天津 300130; 2.河北工業大學天津市電子材料與器件重點實驗室,天津 300130)
由于銅(Cu)具有低電阻率和良好的抗電遷移能力,因此在微電子應用中,銅已經替代鋁成為主要的互連金屬材料。1997年IBM公司提出了雙大馬士革工藝,采用雙金屬鑲嵌技術來制造銅互連,化學機械拋光(CMP)應運而生。它通過機械作用和化學溶解的雙重作用來消除晶圓表面剩余的銅,進而實現局部及全局平坦化[1]。在整個CMP過程中,拋光液的化學腐蝕、鈍化膜的生長以及拋光機與磨料提供的機械研磨三者之間協同作用,可以保證被拋晶圓表面平坦化。
目前,為避免高壓力機械作用造成介質材料的損傷,銅互連CMP技術更多地依賴于化學溶解,主要涉及拋光液各組分與銅基體的相互反應。銅CMP用拋光液通常包含氧化劑、配位劑、抑制劑和磨料顆粒。被拋晶圓表面凸處的銅需要較快去除,而凹處的銅需要保護,氧化劑可以令銅表面生成硬度較低的氧化物鈍化層(包括CuO、Cu2O),容易被機械作用磨除,但是氧化物鈍化層是不連續且多孔的,不能有效抑制銅膜的化學腐蝕[2]。于是人們向拋光液中引入了抑制劑,它通過與銅反應生成鈍化膜并吸附在金屬表面,從而減少凹處的化學溶解(即降低凹處的去除速率),提高凹處與凸處的去除率差異,以獲得全局平坦化和低缺陷的銅表面。
在銅CMP過程中最有效的有機抑制劑是含有N、S、O、P等雜原子以及芳香環的化合物,這些分子可以通過雜原子的孤對電子對(Lp)供給金屬表面未占據的d軌道或氧化膜表面的吸附位點以形成配位共價鍵,或者通過有機化合物接受金屬表面的自由電子形成配位鍵而吸附在金屬表面,是優異的腐蝕抑制劑[3]。
對于抑制劑的研究,主要通過電化學、表面形態學、光譜學等實驗手段研究其吸附類型和特性[4],然而這些方法只能研究抑制劑在銅表面的吸附行為,不能在微觀層面解釋抑制劑在銅表面的吸附機理。因此,越來越多的學者通過分子動力學方法來闡釋抑制劑在銅表面的作用機理,結合量子化學計算,探究抑制劑分子結構與緩蝕效率的關系(即構效關系)[5]。
本文總結了銅互連CMP中抑制劑作用機理的研究手段及研究進展,再根據實驗和量子化學計算來從不同角度解釋抑制劑在銅表面的吸附機理,最后對抑制劑未來的重點研究方向和研究手段進行了展望。
1 抑制劑對銅抑制機理的實驗研究進展
通常根據有機抑制劑分子在銅表面形成物理和/或化學吸附的膜來解釋抑制機理,而抑制性能在很大程度上取決于抑制劑與銅表面之間的相互作用[6]。腐蝕抑制性能主要通過實驗的手段評估,例如質量損失評估、電化學實驗(包括動電位極化、電化學阻抗譜等)、表面形態分析以及吸附等溫線擬合,得到抑制劑分子在金屬表面反應的基本特征。
關于有機雜環化合物BTA(苯并三唑,其分子結構如圖1所示),Y.X.Miao等[7]提出了2種吸附機制:一種是BTA分子直接與Cu2O反應而形成Cu(I)BTA,如式(1)所示;另一種是反應生成Cu(II)BTA配合物沉積在銅表面上。
圖1 BTA的分子結構Figure 1 Molecular structure of BTA

溶液的pH決定了BTA分子的存在形式,進而影響其腐蝕抑制性能[8]。在酸性溶液中,BTA主要以質子化物質BTAH+的形式存在,這種物質在銅表面的化學吸附較弱。在中性、堿性溶液中,BTA的腐蝕抑制機理可概括為:首先,BTA分子通過N7原子與銅表面原子的sp軌道雜化而發生頂位吸附;其次,氧化劑的存在或陽極電位的作用導致銅離子化溶解所產生的Cu2+與BTA反應而形成Cu–BTA配合物,該反應主要是由于BTA三唑環上其中一個或兩個氮原子(N8、N9)與銅發生相互作用。
D.Yin 等[9]認為在堿性環境中,BTA將以BTA?離子形式存在,由于靜電吸附作用,BTA?容易以化學吸附的形式吸附在銅表面和銅氧化而成的Cu(I,II)(即Cu2O和CuO)表面,其吸附結構如圖2所示。
圖2 Cu–BTA在Cu2O上的吸附結構[9]Figure 2 Adsorption structure of Cu–BTA on Cu2O [9]
探究抑制劑在銅表面的抑制機理不僅要考慮抑制劑分子本身的物理化學性質,還要考慮銅表面的性質和狀態。G.Xue等[10]最先通過實驗觀察到,純凈(未氧化)的銅表面形成BTA鈍化膜的能力比正常的銅表面更強,速度更快。A.Kokalj等[11]對比了BTA在CuO和Cu2O表面的吸附行為,發現BTA在Cu2O表面的吸附速率更高。B.J.Cho等[12]進一步研究了銅表面狀態對Cu–BTA配合物形成的影響,采用接觸角測量、電化學阻抗譜(EIS)、X射線光電子能譜(XPS)等手段對BTA處理的3種銅表面──原樣銅、經乙酸處理的銅以及經乙酸和H2O2處理的銅進行了表征,并發現與其他表面相比,經乙酸處理的銅具有更大的接觸角和更高的極化電阻。圖3顯示了不同處理后銅表面的XPS O 1s光譜以及不同處理后銅表面4種氧化物的原子分數。結果表明:BTA更容易吸附在乙酸處理后的純銅表面,其次是以Cu2O為主的原樣銅表面,再次是以乙酸和H2O2處理后形成的以CuO為主的銅表面。
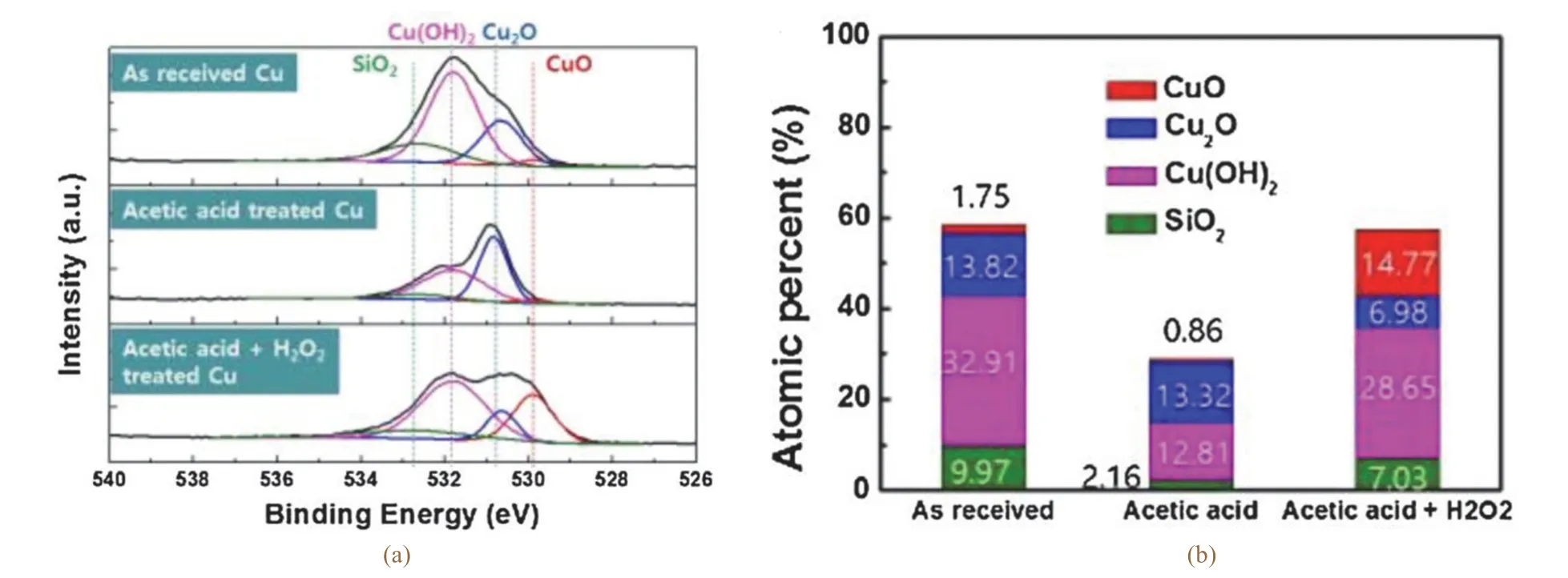
圖3 不同處理后銅表面XPS O1s光譜(a)以及4種氧化物的原子分數(b)[12]Figure 3 XPS O1s spectra (a) of copper surface after different treatments, and atomic fractions of four oxides thereon (b) [12]
B.H.Gao等[13]探究了BTA在不同氧化程度的銅表面的吸附狀態。對未被氧化的銅表面的BTA層進行FTIR(傅里葉變換紅外光譜)分析,結果表明BTA層中的N─H鍵相對減少,即部分BTA分子中的N─H鍵發生了斷裂,與銅表面之間形成了Cu─N鍵。XPS證實了銅表面相鄰的BTA分子與Cu結合形成Cu─N鍵,即銅表面存在一層通過化學吸附而形成的Cu–BTA鈍化膜。經過氧化后的銅會在其表面形成較厚的Cu(II)–BTA結構。
H.Y.Ryu等[14]在研究中發現與BTA同濃度的MBTA(5-甲基苯并三唑)顯示出更好的腐蝕抑制效果,通過EIS發現極化電阻與抑制劑濃度直接相關,并且添加3 mmol/L MBTA顯示出與添加10 mmol/L BTA時相同的極化電阻。換言之,3 mmol/L MBTA與10 mmol/L BTA的腐蝕抑制效率相同。這是由于MBTA中甲基的存在使得三唑環中氮原子的孤對電子親核反應增強。
圖4 MBTA的分子結構Figure 4 Molecular structure of MBTA
BTA在銅表面生成的Cu–BTA鈍化膜足夠致密,需要使用較大的機械力才能得到高的銅拋光速率,因而會導致銅互連的結構性損傷[15]。此外,在銅表面的疏水性BTA使顆粒去除和干燥困難,不僅影響CMP后的清洗工藝,而且影響器件的穩定性和時間依賴性介電擊穿(TDDB)的可靠性。因此,L.Jiang等[16]提出以TAZ(即1,2,4-三唑)代替BTA,通過電化學測試、XPS、靜態腐蝕速率試驗、動態拋光試驗等手段探究了TAZ的腐蝕抑制效果。如圖5所示,隨著TAZ的濃度增加,腐蝕電位正移,腐蝕電流密度減小,表明直接形成了有效的Cu–TAZ鈍化膜;其次,以式(2)所示的反應生成的Cu–TAZ配合物沉積在氧化銅表面形成弱鈍化膜,作為有益的補充。Cu–TAZ鈍化膜較薄,親水性的Cu–TAZ配合物在CMP后的清洗過程中更容易被去除。
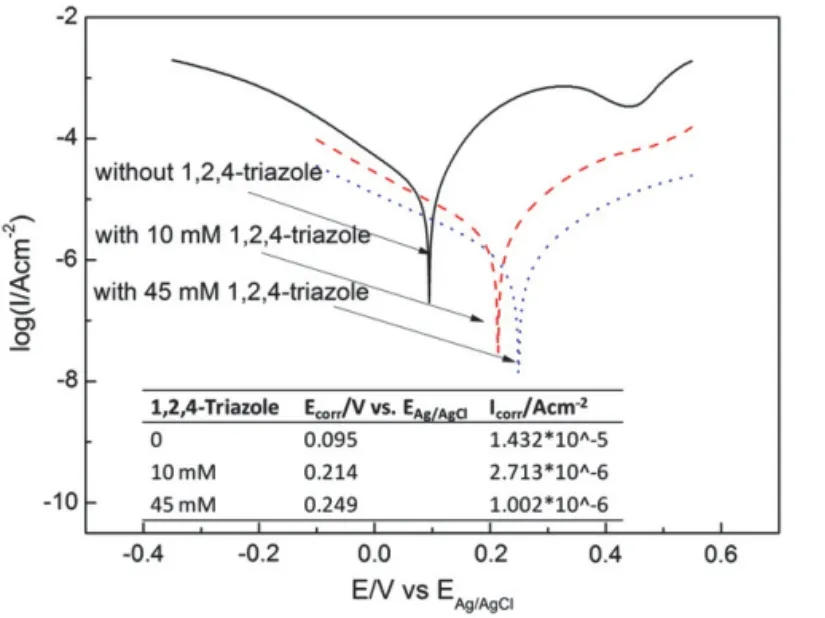
圖5 在含不同TAZ濃度的25 mmol/L Na2SO4 + 1.0% H2O2溶液中銅電極的動電位極化曲線[16]Figure 5 Potentiodynamic polarization curves of Cu electrode in 25 mmol/L Na2SO4 + 1.0% H2O2 solution with different concentrations of TAZ [16]
不同抑制劑在銅表面的吸附類型不同。李靜等[17]進行了TAZ分子等溫線擬合,并計算出其吸附吉布斯自由能= ?25.53 kJ/mol。他們認為TAZ在銅表面的吸附方式為混合吸附,既存在物理吸附膜Cu:(1,2,4-TAH)ads,又伴隨化學反應所形成的聚合膜Cu(1,2,4-TA)2。
pH也會影響TAZ的存在形式,進而影響其吸附性質(如圖6所示)。W.Q.Zhang等[18]根據前人[16-17]的結論,進一步提出在堿性條件下,TAZ在銅表面的物理吸附是由1,2,4-TAH中性分子與金屬表面范德華力的相互作用決定的,化學吸附主要是由1,2,4-TAH分子和1,2,4-TA?陰離子引起的。
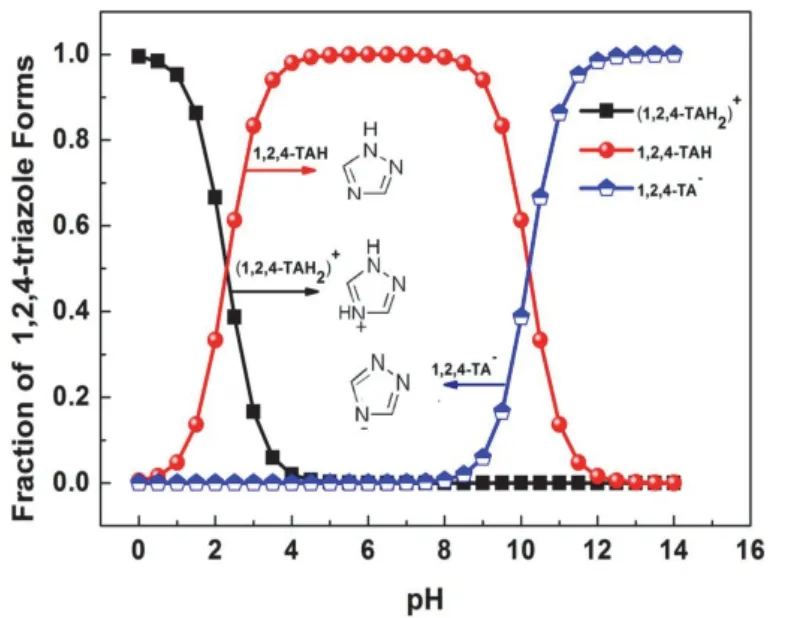
圖6 不同pH下TAZ的存在形式[18]Figure 6 Forms of TAZ at different pH values [18]
根據B.J.Cho等[12]的實驗結果可以得出抑制劑分子的吸附行為與銅表面狀態有關的結論。于是Q.Wang等[19]研究了TAZ在不同氧化狀態的銅表面的吸附速率,結果表明檸檬酸和H2O2處理的銅表面具有較小的接觸角(由于形成了親水性的Cu–TAZ配合物)和較高的極化電阻,這意味著TAZ更容易吸附在檸檬酸和H2O2處理過的銅表面。
L.J.Hu等[20]通過電化學阻抗譜研究發現:新型抑制劑2,2′-[(甲基-1H-苯并三唑-1-基)甲基]亞氨基雙乙醇(TT-LYK)在銅表面有鈍化情況,并用XPS分析了銅表面的Cu(I)–TT-LYK鈍化膜。隨后,J.K.Zhou等[21]設計了相似的實驗來探究含TT-LYK與TAZ的拋光液對銅的腐蝕抑制性,擬合吸附等溫線和計算吉布斯自由能后發現TAZ與TT-LYK分子在銅表面的吸附都是以物理吸附為主導的混合吸附,并猜測TT-LYK在銅表面的鈍化過程包含2個步驟:先是Cu–TT-LYK鈍化膜的生長,然后是Cu–TT-LYK配合物的再沉積。
TAZ和TT-LYK均為唑類抑制劑,相似的結構往往會有相似的性質。如圖7所示,在含有2%(質量分數)甘氨酸和30 mL/L雙氧水的拋光液中同時使用TAZ和TT-LYK抑制劑時,處理后銅表面的XPS譜中N1s的峰值與使用單一抑制劑時相比明顯增加。電化學阻抗譜分析的結果表明2種抑制劑存在協同作用,能在銅表面形成穩定的雙層鈍化膜結構[21]。
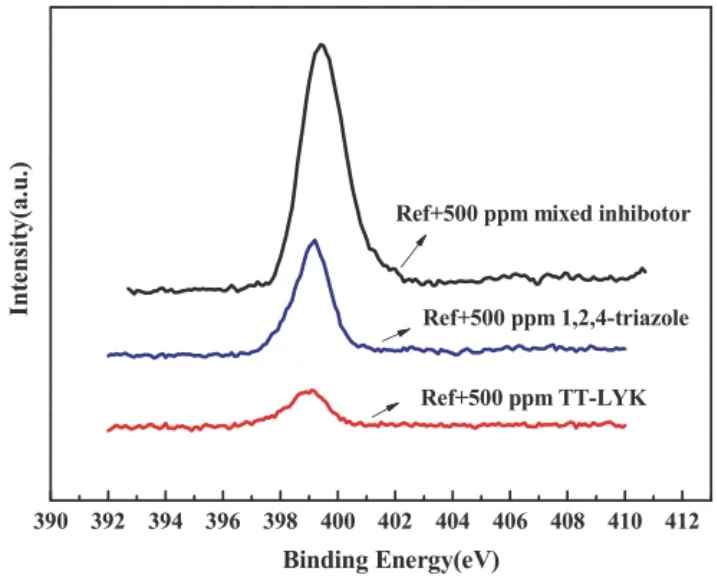
圖7 不同抑制劑處理后銅表面的XPS N1S譜[21]Figure 7 XPS N1s spectra of copper surfaces processed in solutions with different inhibitors [21]
2 抑制劑對銅腐蝕抑制機理的量子化學計算研究進展
由于上述提到的由實驗方法得到的抑制劑在銅表面的作用機理(包括吸附類型、鈍化過程、鈍化膜組成等)并不能詳細闡明抑制劑在金屬表面如何發生反應以及相互作用機理等問題,近年來有學者提出抑制劑分子的有效性與其分子結構有關。而為了更好地理解腐蝕抑制行為,有必要對抑制劑的分子結構進行更系統的研究,如電荷轉移、反應位點數量以及與金屬表面相互作用的類型,等等[22]。為尋求突破,很多研究人員結合量子化學和分子動力學模擬以尋求深層次的機理解釋,預測抑制劑分子的抑制性能[23]。
自1971年J.Vosta與J.Eliásek[24]應用量子化學計算的方法得到吡啶衍生物的前線分子軌道(MO)能量,發現抑制劑分子結構與抑制效率的關系之后,越來越多的研究致力于使用量子化學方法論證抑制劑分子與金屬表面的相互作用機理,并用專門的量子化學方法和分子建模計算軟件(如Materials Studio、Vesta等)研究抑制劑分子特性對抑制效率的影響。DFT(密度泛函理論)被用于計算抑制劑的量子化學參數,包括Fukui指數、前線分子軌道能和分子靜電勢(ESP),這些量子化學參數不僅能根據反應指數和電子、分子特性準確預測抑制劑的抑制效果,還能描述抑制劑結構對抑制效率的影響[25]。
2.1 Fukui指數
Fukui指數用于確定分子活性位點,預測反應中心以及親核或親電性質。在外部勢場(Vr)為常數的條件下,應用有限差分近似后,Fukui函數 ()fr→如(3)和(4)所示。

A.Lesar等[26]采用B3LYP方法和6-31G(d,p)基組,計算了TAZ和3種氨基-TAZ異構體的量子化學參數。Fukui指數計算表明3-氨基-1,2,4-三唑(3-ATA)有2個親電中心(N2、N6)和2個親核攻擊中心(C5、N2),說明親電攻擊優先發生在N2和N6位點,而C5和N2中心是親核攻擊的首選。
S.Y.Tian等[27]根據Fukui指數分析了抑制劑1H-苯并三唑甲酸(CBT)的反應中心。在親核攻擊中,咪唑環上的N17、羧基上的O12是主要的反應位點。氮原子N17和氧原子O12接受銅表面電子對后,雜化形成化學鍵,使CBT吸附在銅表面而阻止腐蝕。
2.2 ESP
分子靜電勢是指核與電子圍繞分子產生的空間型靜電電位分布,用于分析和預測抑制劑分子的反應活性。任意點r→的靜電勢在分子周圍的空間中可以表示成式(5)。

其中ZA是位于處的原子核A的電荷,是分子的電子密度,是特定區域的靜電勢,取決于核或電子在該區域的作用所占的主導地位[28-29]。對比Fukui指數,ESP通過不同顏色展現對應區域靜電勢的大小,使分子表面上靜電勢的分布更加直觀。越接近范德華表面靜電勢最小(大)點的原子就越容易發生親電(親核)反應。
王亞珍等[30]建立了TAZ的分子靜電勢以預測親電攻擊和親核攻擊的首要吸附位點。如圖8所示,紅色代表與親電攻擊相關的最高負電位區,藍色代表與親核反應相關的最高正電位區,據此可以判斷抑制劑分子在銅表面的吸附位點以及研究CMP后清洗中的解吸附機理。
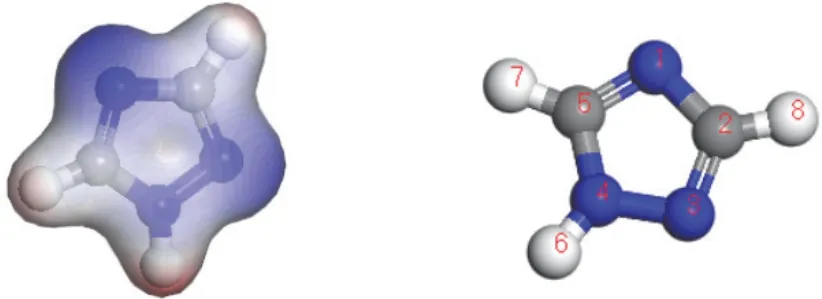
圖8 TAZ的分子靜電勢[30]Figure 8 Molecular electrostatic potential of TAZ [30]
2.3 前線分子軌道能
抑制效率與軌道能量之間有直接的相關性,抑制劑在金屬表面的反應基于抑制劑的電子和金屬表面原子的d軌道之間的作用。與以上研究角度不同的是, 一些學者[31-32]基于前線分子軌道理論,對抑制劑提供和接受電子對的反應位點進行了更加系統的論證。他們發現:抑制劑的性能與其前線分子軌道(MO)和表面吸附位點密切相關,涉及最高占據分子軌道(HOMO)、最低未占據分子軌道(LUMO)和其他參數,其中HOMO(LUMO)是充當電子供體(受體)的軌道。首先,較高的HOMO能量(EHOMO)或較低的LUMO能量(ELUMO)說明電子較易從HOMO轉移到金屬表面或抑制劑分子易于接受來自金屬原子的電子。其次,能隙值?Egap(即ELUMO?EHOMO)同樣是一個重要參數。?Egap降低意味著最外層軌道上捐獻電子提供給金屬的未占據d軌道的能量降低,抑制劑將更容易在金屬表面吸附。
L.J.Hu等[33]指出TT-LYK分子和PO(油酸鉀)分子的前線軌道分布位置如圖9所示,TT-LYK的HOMO主要分布在苯并三唑環的C14和C16上,LUMO主要集中在N═N雙鍵附近。PO的HOMO位點在C═O雙鍵和C─O鍵附近,LUMO集中在C1、C2和C3位置。通過以上量子化學參數,可以明確抑制劑電子分布及反應位點,對后續建立抑制劑在銅表面的吸附模型有很大幫助。
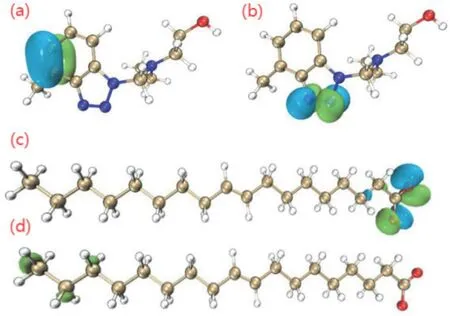
圖9 TT-LYK的HOMO(a)、LUMO(b)以及PO的HOMO(c)、LUMO(d)前線分子軌道密度分布[33]Figure 9 Frontier molecular orbital density distribution of HOMO (a) and LUMO (b) of TT-LYK as well as HOMO (c) and LUMO (d) of PO [33]
3 抑制劑對銅腐蝕抑制機理的分子動力學研究進展
量子化學計算可以判斷抑制劑分子有無明顯吸附,而且通過計算全局(軌道能量等)和局部反應參數(Fukui指數、ESP等)可以比較各類抑制劑的吸附性能,然而量子化學方法缺乏描述抑制劑/金屬表面相互作用的能力[34]。為了從原子尺度了解抑制劑分子中的雜原子在銅表面吸附的過程,通常采用分子動力學方法分析抑制劑分子與金屬表面之間相互作用的動態性質,計算吸附能。利用蒙特卡羅模擬技術,可以通過探索金屬表面的最低吸附位點來確定抑制劑分子的優先吸附位。
S.Q.Sun等[35]建立了咪唑(IMD)、苯并咪唑(BIMD)和2-巰基苯并咪唑(MBIMD)3種抑制劑分子在Cu(III)面上的吸附模型(如圖10所示),其中IMD分子通過氮原子N4,BIMD分子通過氮原子N7吸附在Cu原子的頂位上,而MBIMD分子可以通過N7和S10原子同時吸附在橋位上。這3種抑制劑的相互作用能(?E)分別為?0.54、?0.59和?0.61 eV。因此,與其他2種抑制劑相比,IMD在N4原子頂端的吸附更穩定。
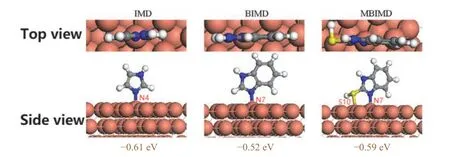
圖10 IMD、BIMD和MBIMD在Cu(111)上吸附的模型[35]Figure 10 Adsorption models of IMD, BIMD, and MBIMD on Cu(111) [35]
T.D.Ma等[36]分析了BTA、TAZ和TT-LYK的HOMO和LUMO分布后,建立了它們在Cu(III)面的吸附模型。從圖11可以看出,這3種抑制劑的N、S和O原子以配位鍵和共價鍵的吸附形式通過平行堆疊的方式覆蓋于銅表面,且TT-LYK的吸附位點更多。他們也計算出了上述3種抑制劑在銅表面的吸附密度,如圖12所示。TT-LYK的吸附密度高于BTA和TAZ,這意味著TT-LYK更容易在銅表面形成致密的鈍化膜。

圖11 BTA、TAZ和TT–LYK在Cu(111)上吸附的模型[36]Figure 11 Adsorption models of BTA, TAZ, and TT-LYK on Cu(111) [36]
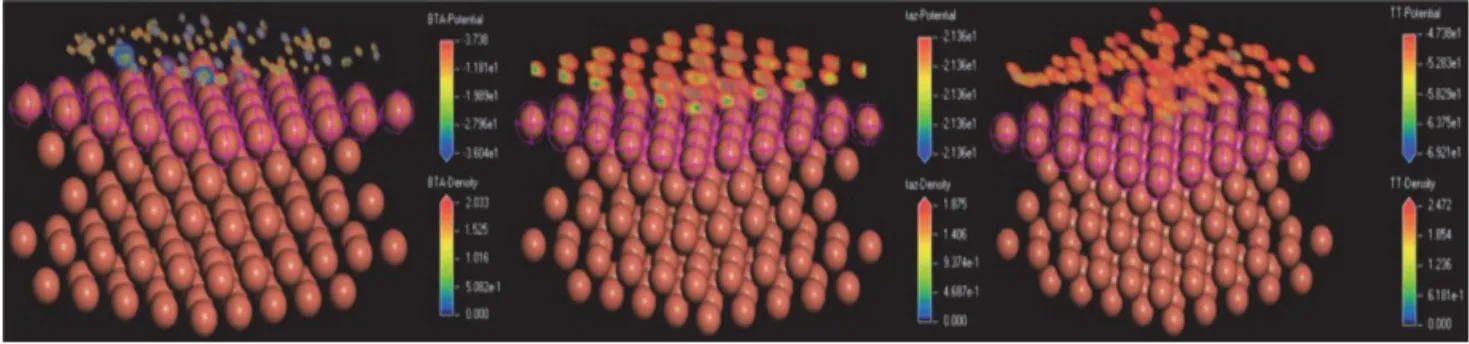
圖12 BTA、TAZ和TT–LYK在Cu(111)上吸附的密度[36]Figure 12 Density of BTA, TAZ, and TT-LYK adsorbed on Cu(111) [36]
D.Kumar等[37]建立了谷氨酸(GLA)、半胱氨酸(CYS)、甘氨酸(GLY)及其衍生物谷胱甘肽(GLT)在銅表面的吸附模型,通過實驗和量子化學計算得知這4種氨基酸類抑制劑的抑制效率排序為:GLT > CYS > GLA > GLY。通過分子動力學模擬計算得到的表面吸附模型如圖13所示。抑制劑分子的N、O和S原子與表面Cu原子之間存在共價鍵,且GLT在銅表面的吸附最強,形成的化學鍵最多,其平行堆疊的吸附結構可提供最大的覆蓋率,從而有效保護了銅表面。
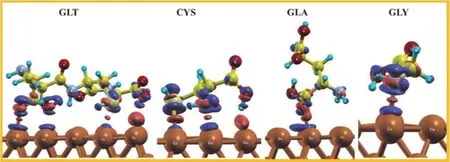
圖13 GLT、CYS、GLA和GLY在Cu(111)上吸附的模型[37]Figure 13 Adsorption model of GLT, CYS, GLA, and GLY on Cu(111) [37]
通過分子動力學研究可以更好地理解抑制劑分子與金屬表面相互作用的機制(包括吸附形式、吸附密度、覆蓋率以及結合能),這是研究抑制劑的關鍵,有助于新型抑制劑的設計和開發。
4 總結與展望
BTA及其衍生物仍然是銅互連CMP中的主要腐蝕抑制劑。由于晶片拋光后表面殘留的BTA會使表面變成疏水性,導致干燥難及堆疊層粘附性差,嚴重影響集成電路性能,因此抑制劑的去除成為銅CMP后清洗的最大難題。而抑制劑的研究重點已經由高效向綠色環保方面過渡。如今以氨基酸類為代表的抑制劑受到廣泛關注,皆因其具有價格低廉、容易獲取等優點,已經在金屬防腐領域應用。混合抑制劑的協同作用可以提高抑制效率,有助于改善CMP后銅表面質量,解決單一抑制劑抑制效率的不足。不同抑制劑的復配和綠色抑制劑的研發可能會成為未來的研究熱點。
量子化學和分子動力學研究可以通過對抑制劑分子在原子層面多角度的計算,探索更深層次的抑制機理,但應輔以電化學等實驗結果的驗證,不可以完全依賴理論計算結果來討論抑制性能。未來關于抑制劑對金屬表面抑制機理的研究應該更全面地使用包括電化學測試、表面分析以及理論計算等在內的各種研究思路和手段。另外,如何從理論層面揭示拋光液中的其他組分(如氧化劑、配位劑)對抑制劑的影響與它們之間的協同效應,以獲得符合要求的CMP材料去除速率和低的靜態腐蝕速率,也將成為研究的重點。
