從化學合成到生物合成
——天然產物全合成新趨勢
2021-11-29 06:40:34張發光曲戈孫周通馬軍安
合成生物學 2021年5期
張發光,曲戈,孫周通,馬軍安
(1 天津大學理學院化學系,合成生物學前沿科學中心,天津 300072;2 中國科學院天津工業生物技術研究所,天津300308)
天然產物是從自然界存在的動物、植物、微生物中分離提取的有機化合物,是生物體在適應環境的漫長進化過程中,為了生存而產生的內源生理活性分子。廣泛而多樣性的生物,制造出千變萬化的天然產物,被認為是大自然賜予人類的瑰寶,也是開發藥物活性分子的重要源泉。例如,8000 年前人類就已種植罌粟用于觀賞和治病,但直到19 世紀初才由德國藥劑師Friedrich Sertürner首次從罌粟中分離出活性天然產物——嗎啡(morphine),這一創舉成為人類將純單體天然化合物用作藥物的里程碑性標志(圖1)[1-2]。人類利用柳樹皮鎮痛退燒也有數千年的歷史,1828 年德國藥物學家Johann Buchner首次從中提取分離出活性成分水楊苷(salicin),逐漸揭開其神奇功效背后的面紗;1838年意大利化學家Raffaele Piria確定了水楊酸苷的結構,并經水解和氧化制得水楊酸(salicylic acid),但因其對咽喉和胃腸刺激劇烈而無法用于臨床治療;1852 年法國化學家Charles Gerhardt將水楊酸鈉與乙酰氯進行反應,首次報道了乙酰水楊酸(acetylsalicylic acid)的合成,但對其功效未進行深入研究;在此基礎上,1897 年德國拜耳公司 Arthur Eichengrün 和 Felix Hoffmann 等重新制備了乙酰水楊酸,發現其獨特的鎮痛退燒功效并申請專利,隨后拜耳公司將其命名為阿司匹林(aspirin)推向市場,開創了人類將天然產物類似物(或衍生物)作為藥物的先河(圖1)[3-4]。
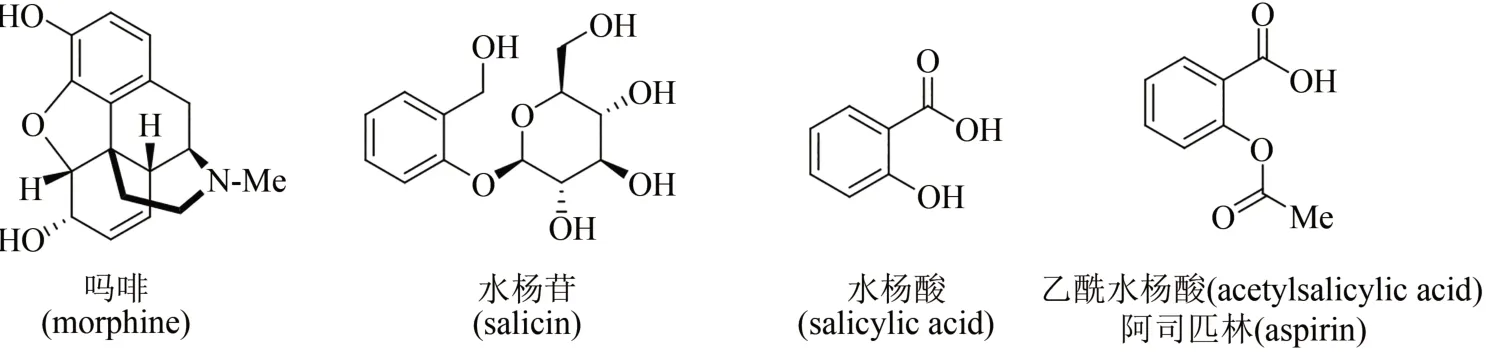
圖1 嗎啡、水楊苷、水楊酸及乙酰水楊酸化學結構Fig.1 Chemical structures of morphine,salicin,salicylic acid,and acetylsalicylic acid
進入20 世紀之后,天然產物化學研究獲得快速發展,大量天然產物從各類動物、植物、海洋生物和微生物中被提取分離和成功鑒定,很多高生理活性分子被用作治療疾病的藥物[5-6]。據統計,目前市場上超過40%的小分子藥物來源于天然產物及其類似物[7-9],例如藥物化合物庫中的明星分子(圖2),有源于植物的奎寧(quinine)和青蒿素(artemisinin),是治療瘧疾的特效藥物;源于植物的紫杉醇(taxol)、長春堿(vinblastine)及長春新堿(vincristine),是廣譜抗癌藥物;源于微生物的青霉素(penicillin)、鏈霉素(streptomycin)、萬古霉素(vancomycin)等,是治療細菌感染類疾病的特效抗生素藥物;而源于微生物的洛伐他汀(lovastatin)則能有效控制膽固醇,為高血脂患者帶來福音;源于植物的二甲雙胍(metformin)及源于哺乳動物的胰島素(insulin),被用于治療和控制糖尿病;源于海洋軟體動物的軟海綿素B(halichondrin B),對一些惡性腫瘤具有神奇療效(圖2)。除此之外,很多天然產物及其類似物還被開發出來,已被廣泛應用于香料、染料、食品添加劑、農藥和獸藥等領域。因此天然產物無處不在,與人類生活密切相關。
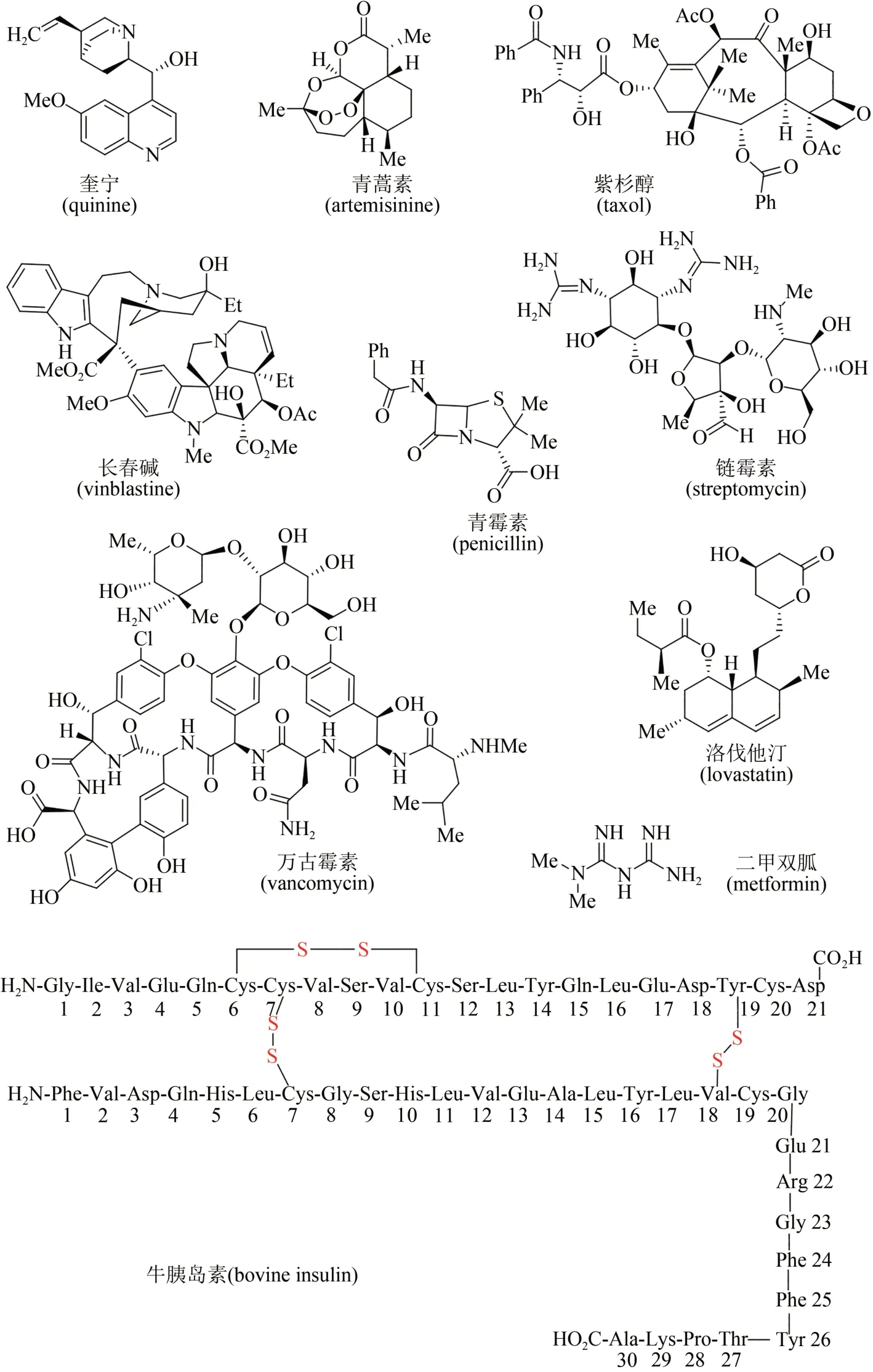
圖2 天然產物中的部分明星藥物分子結構Fig.2 Molecular structures of several star natural products as pharmaceuticals
1 天然產物化學全合成
盡管天然產物具有很好的生物活性,但其在生物體內含量非常低,通過提取分離進行大量制備存在很大難度,從而嚴重制約其開發和利用。1828 年德國化學家Friedrich W?hler 采用無機原料和試劑探索化學反應,在實驗室成功制備出尿素(urea),開創了化學方法獲取有機分子的新時代[10]。1845 年 Hermann Kolbe 用元素碳制備出乙酸(acetic acid),首次提出“合成”(synthesis)這一概念,意指將化學原料通過組裝反應制得產物的過程[11];1869 年 Carl Gr?be 和 Carl Liebermann以蒽為原料經過氧化反應,合成出天然橙紅色染料茜紅素(alizarin)[12];1878 年 Adolf von Baeyer利用鄰硝基苯甲醛與丙酮反應,合成出天然產物靛藍(indigo)[13];而19 世紀最引人矚目的全合成工作,當屬1890 年Emil Fischer 完成的含有5 個相連碳立體中心的葡萄糖(glucose)分子[14]。這些開拓性研究揭開了天然產物化學全合成的新篇章。
進入20 世紀,天然產物全合成領域涌現一批有機化學大師,他們設計發展了各種各樣的化學試劑和有機轉化反應,成功實現眾多天然產物的化學全合成。例如20 世紀初,德國化學家Richard Willst?tter 和英國化學家 Robert Robinson 采用不同的工藝路線,完成了托品酮(tropinone)的全合成[15];到了 20 年代,德國 Hans Fischer 教授實現了血紅素分子(haemin)的全合成[16];30 年代美國化學家Werner E. Bachmann 對雌甾酮分子(equilenin)的全合成率先獲得成功[17]。在隨后的40 多年里,借助于新的分析檢測技術,天然產物全合成更取得了突破性發展,例如美國有機合成大師Robert B.Woodward 與其合作者,先后完成了奎寧(quinine)[18]、膽固醇(cholesterol)[19]、可的松(cortisone)[20]、馬錢子堿(strychnine)[21]、葉綠素 α(chlorophyll α)[22]、頭孢菌素 C(cephalosporin C)[23]及紅霉素A(erythromycin A)[24]等明星分子的全合成;并且在全合成維生素B12(vitamin B12)[25]實驗過程中,發現電環化協同反應現象,與量子化學家Roald Hoffmann 一起建立了分子軌道守恒(the conservation of molecular orbital symmetry)理論[26]。另一位有機合成大師Elias J.Corey除了完成煙曲霉素(fumagillin)[27]、紅霉內酯 B(erythronolide B)[28]、秋水仙堿(colchicine)[29]、喜樹堿(camptothecin)[30]、前列腺素 F (prostaglandin F)[31]、番紅霉素 A(saframycin A)[32]、抗瘧霉素(aplasmomycin)[33]等眾多天然產物的全合成之外,還提出了逆合成分析(retrosynthesis analysis)策略[34],即運用有機反應邏輯,對目標分子進行合理的解構,反推出起始原料和關鍵反應節點,指導設計出復雜分子的全合成路線。
在這期間我國化學家的全合成工作同樣引人矚目:1965 年中國科學院上海生物化學研究所和上海有機化學研究所與北京大學化學系歷時7年完成了牛胰島素(insulin bovine)的全合成,獲得晶體并確定了其分子結構,這是世界上首次以氨基酸為原料、通過化學反應人工制備出蛋白質;經過生物活性測試,合成的結晶牛胰島素和天然牛胰島素具有同樣的生物活性[35-37]。1981 年中國科學院的4個研究所(上海生物化學研究所、上海細胞生物學研究所、上海有機化學研究所、生物物理研究所)和北京大學等單位,利用化學和酶促相結合的方法,首次人工合成了76 個核苷酸的整分子酵母丙氨酸轉移核糖核酸(yeast alanine transfer RNA),其結構與天然分子完全相同,并具有較高的丙氨酸接受和轉移活性[38-39]。1983—1984 年中國科學院上海有機化學研究所周維善院士團隊以手性香茅醛為原料,經過20 步有機反應,實現了抗瘧藥物青蒿素(artemisinin)分子的全合成[40-41]。
20 世紀90 年代至今,有機化學家進一步完成了許多高藥物活性天然產物的全合成。例如1994年美國化學家Kyriacos Nicolau 和Robert Holton 兩個團隊同年攻克抗癌明星藥物紫杉醇分子的全合成(圖 3)[42-48],其中 Robert Holton 以天然氧化綠葉烯為原料,采用線性策略,經過41 步反應,以不到3%總收率得到紫杉醇;除此之外,化學家采用其他原料和策略還發展了多條不同的全合成路線,但都因為合成步驟太多,工藝路線過長,反應條件苛刻,總收率均低于1%,而無法實現規模化放大生產。1981 年法國科學家Pierre Potier 從紫杉莖葉中分離得到一種高含量化合物——10-脫乙酰基巴卡丁-Ⅲ(10-DAB),具有與紫杉醇相似的四并環骨架。隨后開發出四步合成轉化法,將10-脫乙酰基巴卡丁-Ⅲ以80%總收率轉化為紫杉醇,并在美國施貴寶公司實現了工業化生產。1999 年Kyriacos Nicolau、David Evans 和 Dale Boger 三個課題組幾乎同時報道了萬古霉素的全合成工作[49-51];進入21 世紀之后,英國劍橋大學Steven Ley 和其合作者率先報道了雷帕霉素(rapamycin)的全合成[52];美籍日裔化學家Yoshito Kishi先后完成了海洋天然產物海葵毒素(palytoxin)[53]、河豚毒素(tetrodotoxin)[54]、軟海綿素B(halichondrin B)[55]等的全合成;2006年日本衛材(Eisa)公司化學家基于軟海綿素B 結構改造,經62 步有機反應合成出了含有19 個碳手性中心的抗癌藥物——甲磺酸艾日布林(eribulin),該藥物在2010 年獲美國FDA批準,是第1個衍生自天然產物而用于轉移性乳腺癌患者獲得總生存期改善的化療單藥[56-58]。
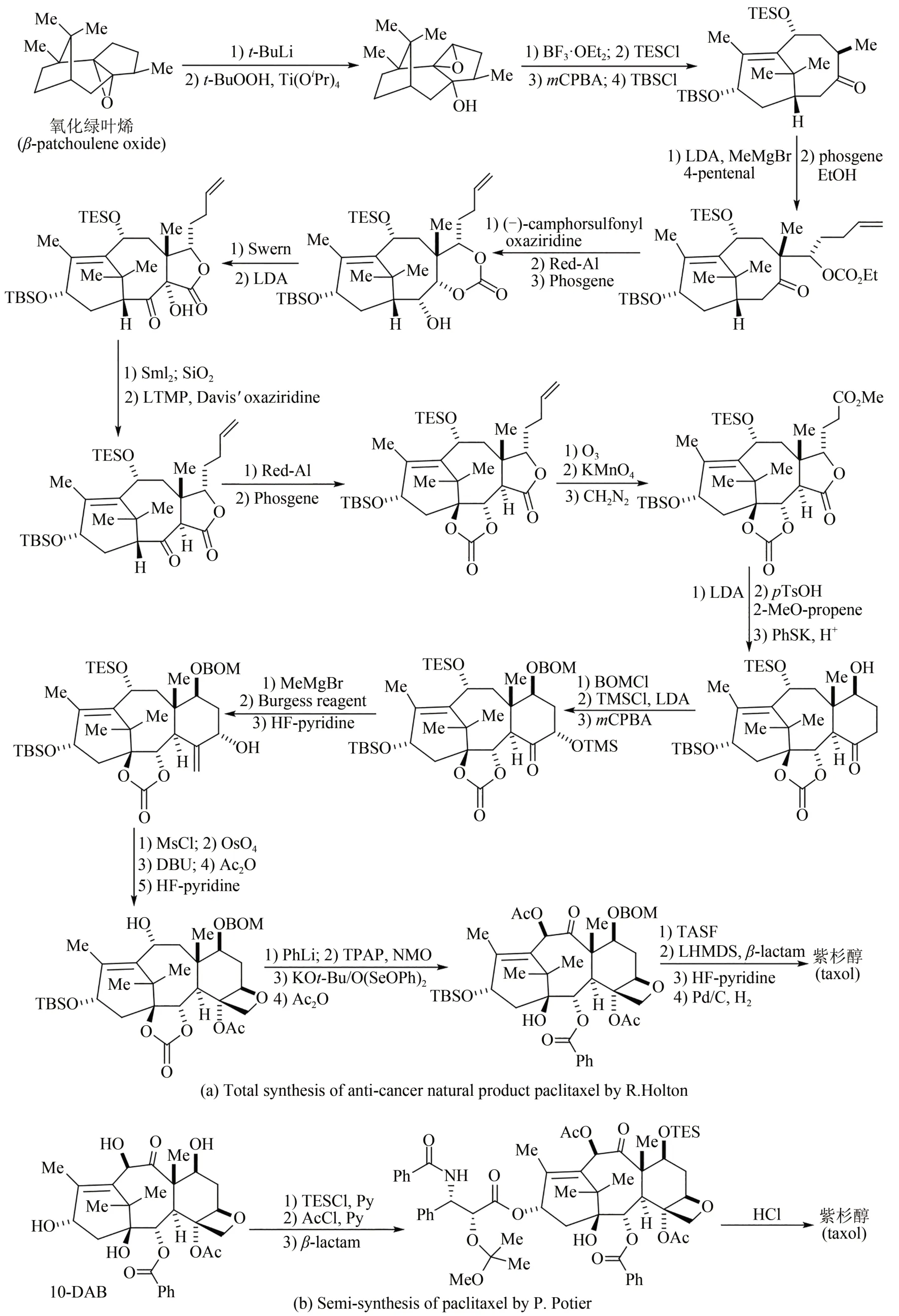
圖3 廣譜抗癌活性天然產物紫杉醇Robert Holton全合成(a)及Pierre Potier半合成路線(b)Fig.3 Total synthesis of the anti-cancer natural product paclitaxel by Robert Holton(a)and its semi-synthesis by the Pierre Potier route(b)
縱覽天然產物的化學全合成,其重要意義主要體現在五個方面:第一,突破了天然來源的限制,從廉價易得的基本原料出發,為獲取天然產物提供了人工化學合成途徑;第二,為復雜天然產物精確化學分子結構的確定,提供了最為直接和有力的證據;第三,以天然產物為先導分子,經過結構改造和修飾,創制出自然界分離不到的活性類似物;第四,促進了化學新試劑、新反應、新方法以及新理論的發展和建立,成為有機合成化學的核心組成部分;第五,天然產物化學全合成還帶動了藥物化學的全面快速發展,也為人類探索和認識生命奧秘提供了有力支撐。盡管如此,天然產物化學全合成也有其局限性,例如對一些結構復雜的高藥理活性分子,化學全合成步驟繁雜,工藝路線長,所用化學試劑種類多,有些反應條件極其苛刻,立體選擇性控制難度高;因此總合成效率非常低,不易大規模工業化制備。天然產物來源于自然界生物體,綠色生物制造方興未艾,為天然產物全合成提供了新的互補性策略。
2 天然產物生物全合成
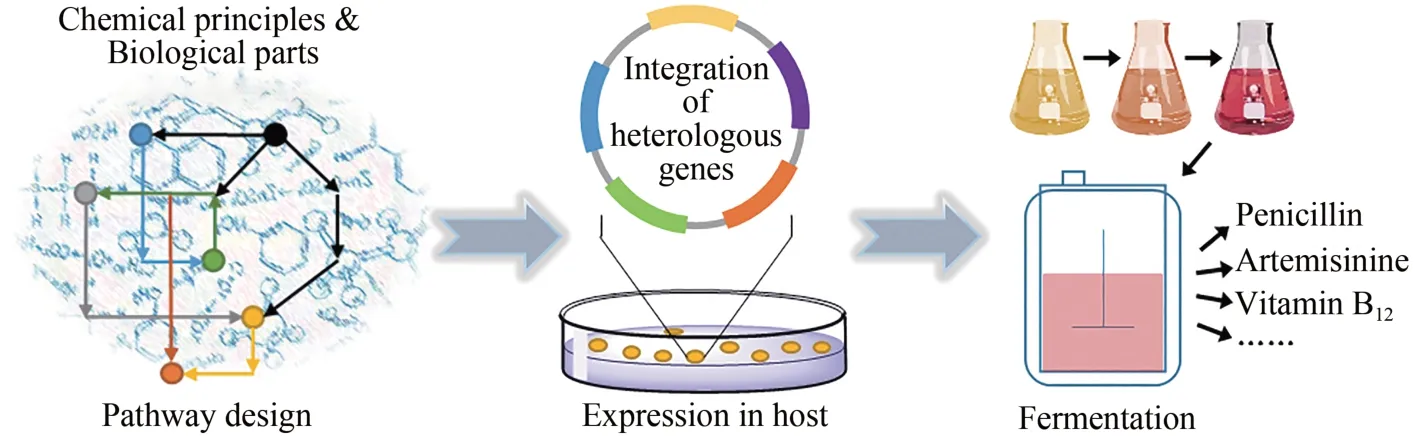
天然產物的化學提取分離與結構鑒定,促進了生物合成研究的發展。早期科學家研究微生物次級代謝天然產物的策略較為單一,主要通過大量篩選尋找能夠分泌具有藥理活性的微生物菌種,然后進行培養和優化研究,并對最佳菌種開展大規模工業化發酵,最后通過提取和精制,完成天然產物生物全合成制備。如今,隨著基因組學、轉錄組學、蛋白組學、代謝組學和生物信息學等多學科交叉領域研究的不斷深入,合成生物學家通過物質/能量代謝及其調控通路的重構,可以在同源或異源微生物細胞中實現天然產物的生物全合成[59-64]。生物制造給天然產物的全合成,帶來了一場影響深遠的變革。在此將通過幾個典型的同源和異源微生物細胞合成途徑重構的例子,分析天然藥物分子的生物全合成。
進行小型農田水利工程的建設管理技術研發時,首先應在工程的信息自動化技術方面投入較多的精力,積極運用計算機網絡技術實現信息管理工作的自動化,對工程管理工作過程中的各項數據信息進行統一的分析和處理,繼而為構建全新的節水型水利工程奠定基礎。其次,在工程水資源的配置方面加大技術研發力度。具體而言,需要按照實際的水利產業結構進行技術規劃,有效將工業化思維融入技術研發工作中,實現對工程管理技術研發階段的系統化劃分。
2.1 天然產物合成的途徑改造與優化
自然界微生物的種類多樣、生長速度快、容易培養和優化等特點。在此基礎上,對微生物次級代謝產物發現及利用促進了天然產物生物全合成領域的逐漸成長。隨著科學和技術的不斷發展,進一步通過對同源微生物細胞的遺傳操作,可以實現次級代謝天然產物的高效調控生物全合成[65-66]。
2.1.1 青霉素生物合成
1928 年英國微生物學家Alexander Fleming 最早發現了青霉菌分泌物——青霉素,但由于當時技術條件的局限,他并沒有分離純化出青霉素。10 年后德國生物化學家Ernst Chain 閱讀了Fleming的研究報道,開始嘗試提純實驗;1941 年他與英國牛津大學病理學家Howard Florey 一起實現了青霉素的分離與純化,并發現其對鏈球菌、白喉桿菌等多種細菌感染疾病的療效。隨后,Florey又在腐爛的甜瓜上發現了一種可供大量提取青霉素的菌種,并使用玉米粉配制出相應的培養液。在這些研究成果推動下,美國制藥企業于1942 年開始對青霉素進行大批量生產;1945 年英國化學家Dorothy Hodgkin 用X 射線衍射法確定了青霉素的分子結構[67]。但直到20世紀80年代,青霉素的生物合成途徑才獲得解析[68-69]。如圖4 所示青霉素G的生物合成途徑:葡萄糖經過代謝后合成3種重要的前體氨基酸,分別為L-纈氨酸(L-Val)、L-半胱氨酸(L-Cys)和L-氨基己二酸(L-AAA),然后經ACV 三肽合成酶縮合形成ACV 三肽,通過異青霉素N 合成酶(ⅠPNS)催化環化合成出異青霉素N(ⅠPN),再經酰基轉移酶AT通過側鏈轉換得到青霉素G。發展至今,青霉素的工業生物全合成已經非常成熟,主要包括三個流程:首先將青霉菌接種到固體培養基上,室溫培養制取青霉菌孢子培養物;隨后將孢子懸浮液接種到帶有滅菌培養基的種子罐中,攪拌發酵培養;最后將發酵液過濾,提取和精制。繼青霉素之后,鏈霉素、氯霉素、土霉素、四環素、金霉素、萬古霉素等抗生素不斷被發現,也都是通過類似流程,實現了大規模工業化生產。
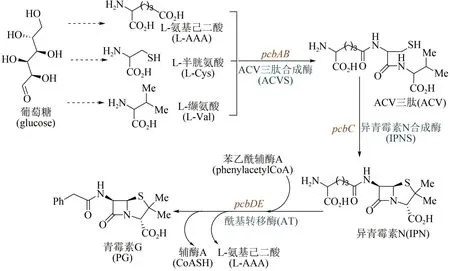
圖4 青霉素生物合成途徑Fig.4 Biosynthetic pathway of penicillin
2.1.2 紅霉素生物合成
作為放線菌來源的聚酮類天然產物,紅霉素屬于大環內酯類抗生素,可用來治療革蘭氏陽性細菌感染。1952 年James McGire 等首次從紅色糖多孢菌(Saccharopolyspora erythraea)的發酵產物中分離得到紅霉素[70]。目前紅霉素的生物體內合成途徑已被探明[71]:首先通過Ⅰ型聚酮合酶(polyketide synthase-Ⅰ, PKS-Ⅰ)多步延伸模塊催化1 分子丙酰輔酶A(propionyl-CoA)和6 分子甲基丙二酸單酰輔酶A[(2S)-methylmalonyl-CoA],合成十四元環中間產物6-脫氧紅霉內酯(6-deoxyerythronolide B,6-dEB);此后,6-dEB 經歷多步修飾,包括P450 羥化酶(EryF)在其大環骨架C6位點進行羥化等反應, 生成紅霉內酯(erythronolide B);再通過兩步糖基轉移反應,即糖基轉移酶EryBV在C3羥基位點連接一個L-碳霉糖,形成3-O-碳霉糖基紅霉內酯(3-O-mycarosyl erythronolide B),以及在此基礎上經糖基轉移酶EryCⅢ在C5 羥基加上一個D-德胺糖,進而形成紅霉素生物合成途徑中第1個具有生物活性的中間產物紅霉素D(Er-D)。接下來,羥化酶EryK 催化紅霉素D 的C12 位點羥化,形成紅霉素C(Er-C),最后經甲基化酶EryG在碳霉糖糖基的C3位點進行甲基化修飾,得到最終產物紅霉素A(Er-A)(圖5)。
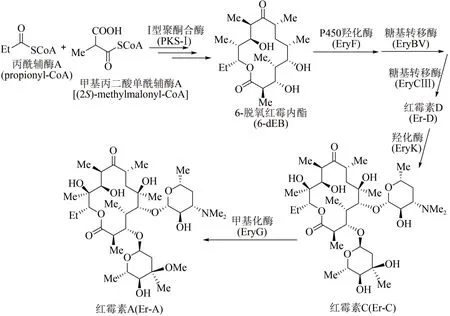
圖5 紅霉素A生物體內全合成路線Fig.5 Biosynthetic pathway of erythromycin A
盡管對于紅霉素在生物體內合成路線研究得比較清楚,但如何提高產量仍具有很大挑戰。中國科學院上海有機化學研究所劉文團隊,運用組合生物合成技術對紅霉素工業用高產菌株進行了針對性的遺傳改良,例如:倍增了紅色糖多孢菌體內PKS 編碼基因,使紅霉素產量提高50%,并使發酵周期縮短1/3[72];通過優化整合到染色體上關鍵羥化酶基因eryK和甲基化酶基因eryG的拷貝數,可以消除副產物B 和C,同時紅霉素A 的產量可以提高30%[73],為紅霉素的工業化生產奠定了基礎。
2.1.3 阿維菌素生物合成
作為農作物保護的高效殺蟲劑,阿維菌素是一種具有十六元環結構的聚酮大環內酯類抗生素,工業上主要采用除蟲鏈霉菌(Streptomyces avermitilis)發酵分離獲得[74]。21 世紀初,阿維菌素的生物合成基因模塊已被科學家全部探明[75-76],其生物體內合成途徑主要包括3 個階段(圖6):①起始單元合成,由L-異亮氨酸(L-Ⅰle)及L-纈氨酸(L-Val)兩條路徑出發,經多步酶促反應分別生成2-甲基丁酰輔酶A(2-methylbutyryl CoA)及異丁酰輔酶A(isobutyryl CoA);②大環內酯骨架合成,在多模塊聚酮合酶PKS 作用下,這兩種起始單元轉化為阿維菌素大環內酯骨架結構;③阿維菌素合成,經多步聚酮后修飾反應,包括環氧化、糖基化、還原及甲基化等,大環內酯骨架轉化為系列阿維菌素化合物[77]。根據其骨架C5、C22、C23 以及C26 位點結構上的差別,阿維菌素由8 個組分構成,即4 個主要組分(A1a、A2a、B1a、B2a)以及4 個次要組分(A1b、A2b、B1b、B2b)。其中B1a 毒性最低、生物活性最強,也是市售阿維菌素農藥的主要殺蟲成分。
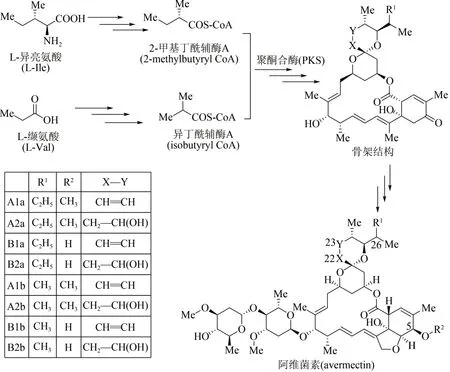
圖6 阿維菌素生物合成途徑Fig.6 Biosynthetic pathway of avermectin
2.2 天然化合物的異源表達與生物制造
自然界動物和植物的多樣性,是眾多天然產物的重要源泉。但由于動植物的結構復雜、代謝更新周期長以及資源局限性等問題,大量獲取這類天然產物難度高。近年來,科學家通過解析天然產物的結構,探索其生化反應機制和合成路線,將相關基因在異源微生物細胞里進行表達和調控,從而完成這類天然產物的生物全合成[79-81]。
2.2.1 細菌合成維生素B12
維生素B12是一種含有三價鈷的多環有機分子,又叫鈷胺素,也是唯一含有金屬元素的維生素。可參與制造骨髓紅細胞,防止惡性貧血,保護大腦神經系統不受破壞。只能從動物的內臟中經人工提取和精制得到,價格極其昂貴。美國哈佛大學Robert Woodward 教授與蘇黎世聯邦理工學院(ETH) Albert Eschenmoser 教授聯合組織了14 個國家的上百位有機化學家,歷時12 年協同攻關,在1973 年成功實現維生素B12的化學全合成[82-83];但由于有機反應步驟多,合成路線太長,根本無法進行大量制備。2018 年,中國科學院天津工業生物技術研究所張大偉團隊,在大腸桿菌中實現了維生素B12的從頭合成(圖7)。首先解析維生素B12好氧合成路徑中鈷螯合與腺苷鈷啉醇酰胺磷酸的合成機理,將來源于5 種細菌中的28 個基因在大腸桿菌細胞中成功異源表達,并按其人工合成途徑劃分為5個模塊進行人工途徑組裝。在克服多基因適配等難題后,最終實現了維生素B12的從頭合成,通過途徑優化和發酵過程調控,產量達到307.00μg/g干細胞菌體,合成菌種發酵周期僅為目前工業生產菌株的1/10,極具工業應用前景[84]。
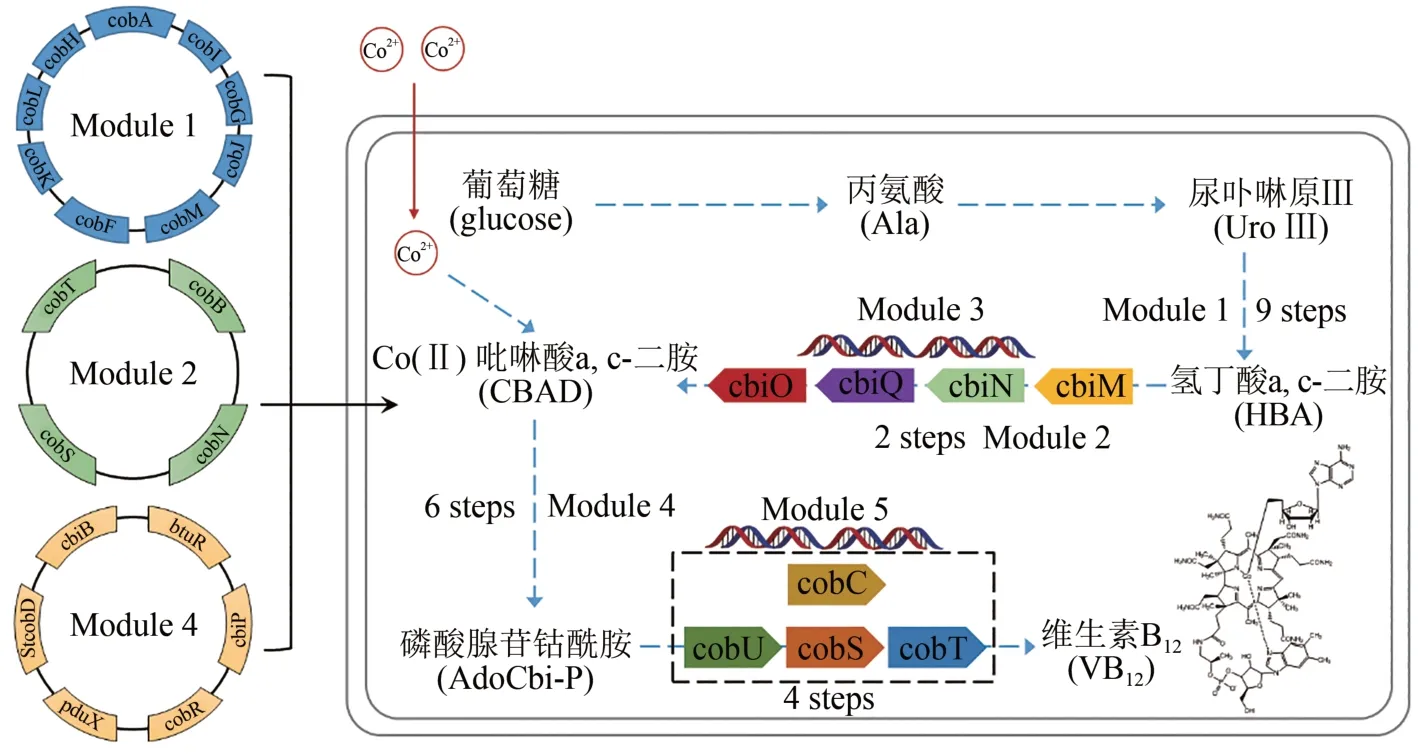
圖7 維生素B12從頭生物合成Fig.7 De novo biosynthesis of vitamin B12
2.2.2 酵母合成莨菪烷堿
來自茄科植物的莨菪烷類生物堿是神經遞質抑制劑,可用于治療神經肌肉疾病,已經被世界衛生組織列為基本藥物。從天然植物獲取這類藥物,受到氣候環境等因素的嚴重制約,而化學全合成路線較長、副產物較多,因此該類化合物的生物合成策略逐漸受到關注[85]。莨菪烷類生物堿的生物合成途徑共涉及13個酶[圖8(a)]:從鳥氨酸開始,經歷鳥氨酸脫羧酶(ODC)→N-甲基轉移酶(PMT)→N-甲基腐胺氧化酶(MPO)→自發環化→Ⅲ型聚酮合酶(PYKS)→托品酮合成酶(CYP82M3)→托品酮還原酶(TRⅠ)歷程形成托品堿;另一方面從苯丙氨酸出發,經歷氨基轉移酶(ArAT4)→苯丙酮酸還原酶(PPAR)→糖基轉移酶(UGT1)形成糖基化苯乳酸,隨后經歷立托林合成酶(LS)→立托林變位酶(CYP80F1)→莨菪醛脫氫酶(HDH)形成莨菪堿,進一步羥化(莨菪堿羥化酶H6H)可生成山莨菪堿及東莨菪堿。西南大學廖志華團隊在莨菪烷類生物堿的生物途徑解析方面做出系列原創性貢獻,先后確立了ODC、PPAR、 UGT1 和 LS 的 酶 學 機 制[86-88]。 2019 年 ,廖志華團隊與中國科學院昆明植物研究所黃勝雄團隊利用轉錄組種內差異表達和種間同源基因分析[89],發現了不同植物來源的3 個Ⅲ型聚酮合酶(AaPYKS、DsPYKS 和AbPYKS),并驗證了其能催化N-甲基吡咯啉陽離子與丙二酰輔酶A 縮合形成三羰基戊二酸,隨后發生自發Mannich反應得到托品烷骨架關鍵前體,揭示了托品烷生物堿生物合成中基本骨架形成的酶學機制[圖8(b)]。近期這兩個團隊進一步發現并鑒定了莨菪堿生物合成途徑中[90],催化莨菪醛生成莨菪堿的關鍵酶——莨菪醛脫氫酶(hyoscyamine dehydrogenase,HDH),蛋白晶體結構及體外酶促反應研究揭示該酶在生理條件下利用NADPH 將莨菪醛還原為莨菪堿[圖8(c)],這些研究標志著以莨菪堿為代表的藥用托品烷類化合物的生物合成途徑得以完整解析,將為莨菪堿的合成生物學異源創制和工業制造提供基礎。
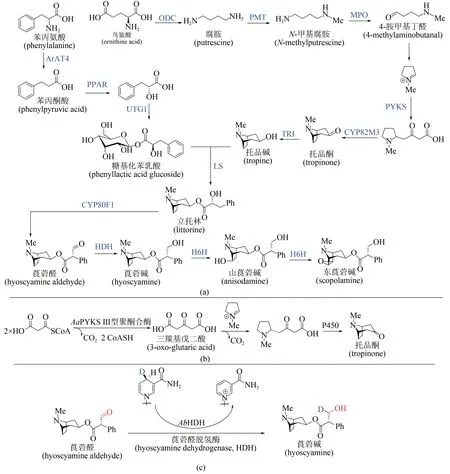
圖8 莨菪烷堿的生物合成機制解析Fig.8 Biosynthetic pathway of tropane alkaloids
來自斯坦福大學的Christina Smolke團隊在酵母中整合多步異源表達基因,通過不同模塊組裝和亞細胞器定位,成功實現了莨菪堿(hyoscyamine)和東莨菪堿(scopolamine)的異源合成[91]。生物合成途徑如圖9 所示:模塊Ⅰ,以天然谷氨酸為前體,通過兩種不同的雙酶途徑,精氨酸酶(Car1)/鳥氨酸脫羧酶(spe1)或精氨酸脫羧酶(AsADC)/胍丁胺脲水解酶(speB)分別合成腐胺;模塊Ⅱ,腐胺經過甲基化(AbPMT1/DsPMT1)、氨氧化(DmMPO1突變體)、自發環化合成N-甲基吡咯啉,再經聚酮合酶(AbPYKS)、 托品酮合成酶(AbCYP82M3)、P450還原酶(AtATR1)和托品酮還原酶(DsTR1)催化合成托品堿;模塊Ⅲ,涉及糖基化苯乳酸的合成途徑,具體包括苯丙氨酸經氨基轉移酶(Aro8/Aro9)、苯丙酮酸還原酶(WfPPR)、糖基轉移酶(AbUGT)的催化,合成糖基化苯乳酸;模塊Ⅳ,托品堿轉運至液泡中,經蛋白質工程改造過的立托林(littorine)合成酶(AdLs)催化,糖基苯乳酸和托品堿轉化為立托林;模塊Ⅴ,立托林轉運至細胞質中再經過異構化(AbCYP80F1/AtATR1)及醛還原酶(DsHDH)催化得到莨菪堿,進一步在雙加氧酶(DsH6H)作用下制得東莨菪堿(圖9)。該工作整合了34個染色體修飾(26 個基因以及8 個突變基因),在不同亞細胞位置上定位了20 多種酶,構建了一個完整的全細胞體系,莨菪堿和東莨菪堿的合成滴度達30 μg/L。該微生物合成平臺可進一步發掘合成新的生物堿衍生物,用于新的藥物開發。
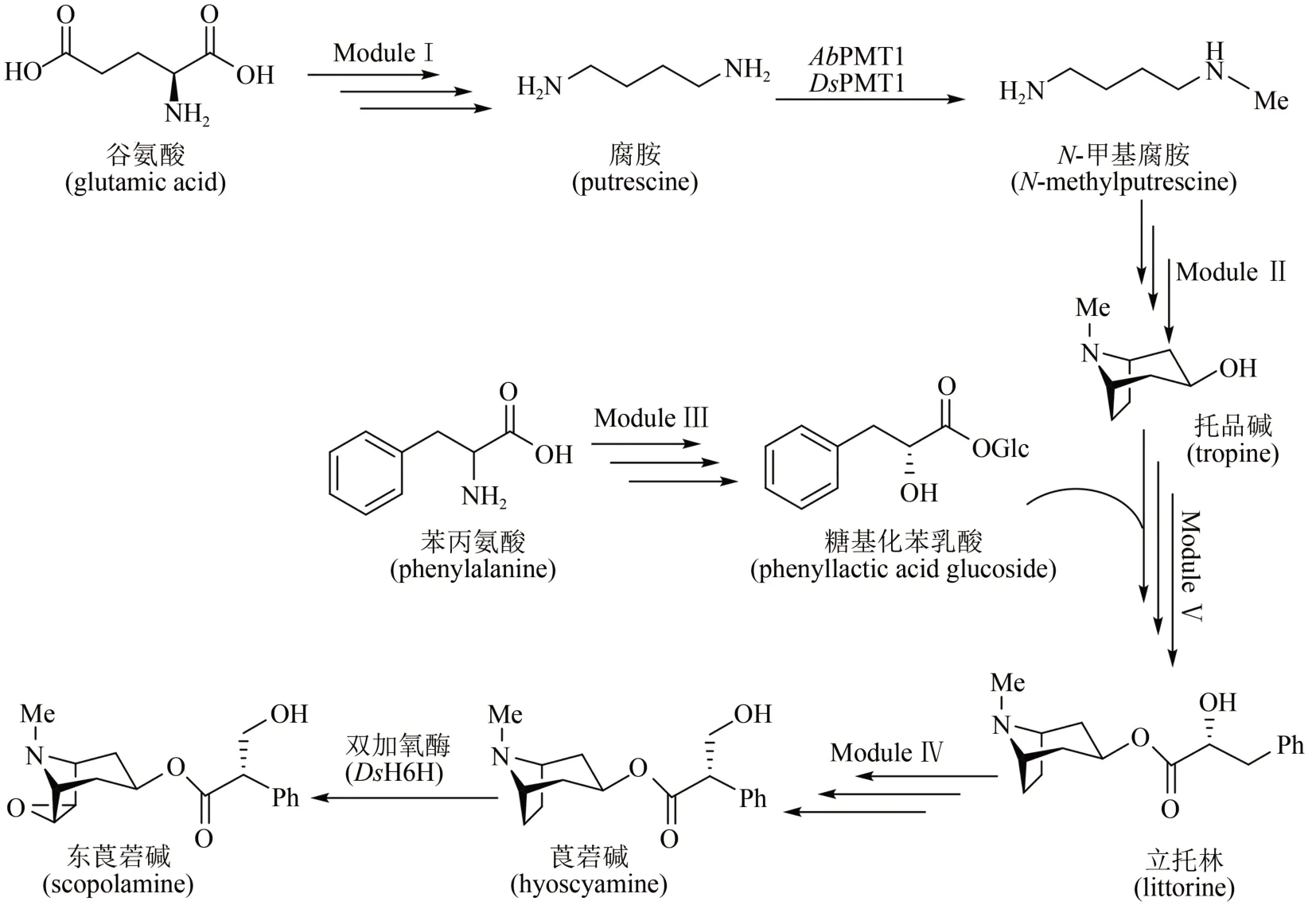
圖9 莨菪烷堿的酵母異源生物合成途徑Fig.9 Heterologous biosynthesis of tropane alkaloids in yeast
3 天然產物生物與化學交叉全合成
如上所述,生物合成策略在天然產物全合成中發揮越來越重要的作用,但仍面臨諸多挑戰性問題,如對一些來源獨特的復雜天然產物,一方面,挖掘生物體內相關基因序列、解析生化反應機制以及特異性遺傳修飾生物合成途徑還存在較大困難;另一方面,將不同來源的天然產物生物合成基因進行重組,在異源微生物體內構建全新的代謝途徑,還存在匹配和耐受性問題;此外,生物酶對底物的專一性要求非常高,天然產物分子骨架的衍生拓展性較差。在這些方面,化學合成可以發揮重要的互補優勢,因而利用生物與化學交叉融合策略制備天然產物逐漸引起關注,成為天然產物及其衍生物全合成的另一個新趨勢[92-93]。
3.1 人源胰島素的生物與化學全合成
胰島素分子的氨基酸序列在20 世紀50 年代已經解析清楚,隨后科學家也嘗試了很多策略用于人工合成胰島素,但由于合成步驟太長、效率太低而無法實現大量工業生產,因此主要來源依然是通過粉碎牛或豬內臟進行提取,每生產1 kg 胰島素則需要超過8 t 動物胰臟。20 世紀60~70 年代,重組DNA 技術取得了重大突破。美國加州大學洛杉磯分校微生物學家Herb Boyer教授團隊通過重組DNA 技術,實現了人源胰島素的全合成:他們采用化學從頭合成的方法,先合成出編碼胰島素A鏈和B 鏈兩條DNA 分子,將這些DNA 插入細菌的基因組使它們能夠合成出胰島素A 鏈和B 鏈分子,再將兩條肽鏈進行分離純化,最后運用化學手段將兩條鏈連接,獲得人源胰島素蛋白分子(圖10)[94-96]。此外,也可以通過大腸桿菌或巴斯德畢赤酵母直接合成單體或胰島素原,進一步經過二硫鍵匹配和酶切合成胰島素[97]。Herb Boyer與風險投資人Robert Swanson一起創立了美國基因泰克(Genentch)生物技術公司,成功實現了人源胰島素的工業化大量生產。
3.2 青蒿素的生物與化學交叉全合成
青蒿素屬于植物萜類天然產物,是20 世紀60~70 年代以屠呦呦教授為代表的我國科學家,從植物青蒿中提取分離出來的具有高效抗瘧活性的有機分子[98]。目前主要從黃花蒿中直接提取,或提取黃花蒿中含量較高的青蒿酸,然后經化學半合成制得。由于受氣候條件和地區環境等因素影響,產量嚴重受限。隨著越來越多關鍵酶基因被表征鑒定,青蒿酸的生物合成途徑已獲得全部解析;通過在微生物細胞中異源表達,進行生物合成青蒿酸引起科學家高度關注。在這方面最具代表性的是美國加州大學伯克利分校Jay Keasling教授實驗室(圖11)[99],他們從乙酰輔酶A 起始,通過甲羥戊酸途徑合成異戊烯基焦磷酸(isopentenyl phosphate, ⅠPP)及其異構物二甲基烯丙基焦磷酸(dimethylallyl diphosphate,DMAPP),然后經法尼基焦磷酸合成酶(farnesyl diphosphate synthase,FPS)催化ⅠPP 聚合生成法尼基焦磷酸(farnesyl diphosphate,FPP);再采用基因工程手段,將關鍵編碼紫穗槐-4,11-二烯合酶(amorpha-4,11-diene synthase,ADS)基因引入大腸桿菌中,實現了由FPP 到紫穗槐-4,11-二烯(amorpha-4,11-diene)的催化過程;隨即又在酵母中表達P450 紫穗槐二烯氧化酶CYP71AV1、還原酶CPR1及細胞色素CYB5等基因,將紫穗槐二烯水解為青蒿醇(artemisinic alcohol),并進一步氧化為青蒿醛(artemisinic aldehyde);再經青蒿醛雙鍵還原酶DBR2 和醛脫氫酶ALDH1 兩步反應,分別將青蒿醛催化生成二氫青蒿醛(dihydroartemisinic aldehyde)以及二氫青蒿酸(dihydro-artemisinic acid,DHAA),發酵產量可以達到25 g/L。隨后以青蒿酸為底物進行后續化學轉化,威爾金森催化劑[RhCl(PPh3)3]催化下可以高達94∶6 的非對映選擇性得到二氫青蒿酸,酯化后使用鉬酸鋰促進的雙氧水歧化反應得到烯丙基過氧化物,進一步在一鍋內發生重排、氧化、成環步驟可生成最終的青蒿素,四步的化學合成總收率可以達到45%。值得注意的是,賽諾菲公司的Turconi 等使用光照反應策略可將二氫青蒿酸以55%的收率轉化為目標青蒿素[100],并可實現370 kg 的放大量生產[圖11(b)]。2015 年,Michael George 等對光照條件下二氫青蒿酸到青蒿素這一關鍵步驟進行了進一步優化[101],他們發現以液態二氧化碳為溶劑可以50%的收率得到目標青蒿素,而使用四氫呋喃/水的混合溶劑體系可進一步提升收率到66%,并可省去煩瑣的分離純化步驟,這些反應條件更加符合綠色化學的發展要求,具有很好的放大應用潛力[圖11(b)]。
3.3 沙弗拉霉素的化學-酶法全合成
雙四氫異喹啉生物堿是一類具有重要抗腫瘤活性的天然產物[102],但化學合成路線步驟煩瑣[103],而單一的生物合成方法效率偏低[104],嚴重制約了該類天然產物的大量獲取及后續活性研究。2018 年,日本東京農工大學Hideaki Oikawa與Hiroki Oguri 課題組首先通過化學方法,巧妙設計并制備出醛類底物;再以磷酸泛素化Pictetpengler 環合酶SfmC 為關鍵酶,催化形成核心的五并環骨架,該反應可在一鍋內同時實現兩個碳-碳鍵、三個碳-氮鍵的構建及硫酯的還原,避免了活性中間體的分離純化,極大提高了合成效率;隨后采用化學方法實現二級胺的甲基化、側鏈水解切斷及氧化官能團轉化。該方法僅需4~5 步反應,即可實現3 種雙四氫異喹啉生物堿(沙弗拉霉素A、Fmoc-保護的沙弗拉霉素Y3 和jorunnamycin A)的全合成,是目前已報道的沙弗拉霉素最短合成路線(圖12)[105]。該研究充分展示了化學-生物交叉融合策略在天然產物全合成中的獨特優勢,也為這類生物堿的規模化制備提供了新的工藝路線。
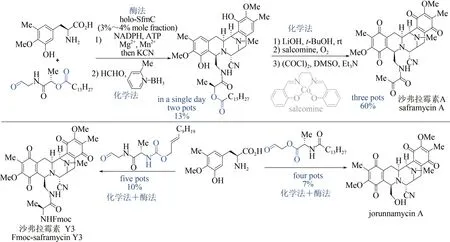
圖12 沙弗拉霉素的化學-酶法全合成Fig.12 Syntheses of saframycins through the chemo-enzymatic catalysis
3.4 嗜氮酮的化學-酶法立體多樣性全合成
嗜氮酮是一類來源于真菌的并環骨架天然產物,具有抗癌、抗病毒、抗炎等多種生物活性,其中僅有的一個四取代碳手性中心對其參與的生理過程發揮重要作用,但立體多樣性合成的空白嚴重限制了該類天然產物的藥用研究。2019 年,美國密歇根大學Alison Narayan 小組通過序列相似性網絡(sequence similarity network,SSN)策略對黃素依賴的單加氧酶進行系統分析,發現了可催化氧化去芳構化反應的一系列新酶,并從中確定了兩種可實現相反對映選擇性的新酶:AzaH(R選擇性)和AfoD(S選擇性)。作者采用化學合成法得到烯酮中間體,通過酶催化的氧化去芳構化反應分別得到兩種絕對構型相反的手性雙環烯酮醇,再借助Knoevenagel 縮合反應高選擇性地實現了三并環骨架的構建,最后成功得到天然產物trichoflectin 的兩個對映異構體,該策略還進一步應用于嗜氮酮同系天然產物deflectin 與lunatoic acid A 的全合成(圖13)[106-107],充分展示了化學-生物結合策略的優勢互補性。
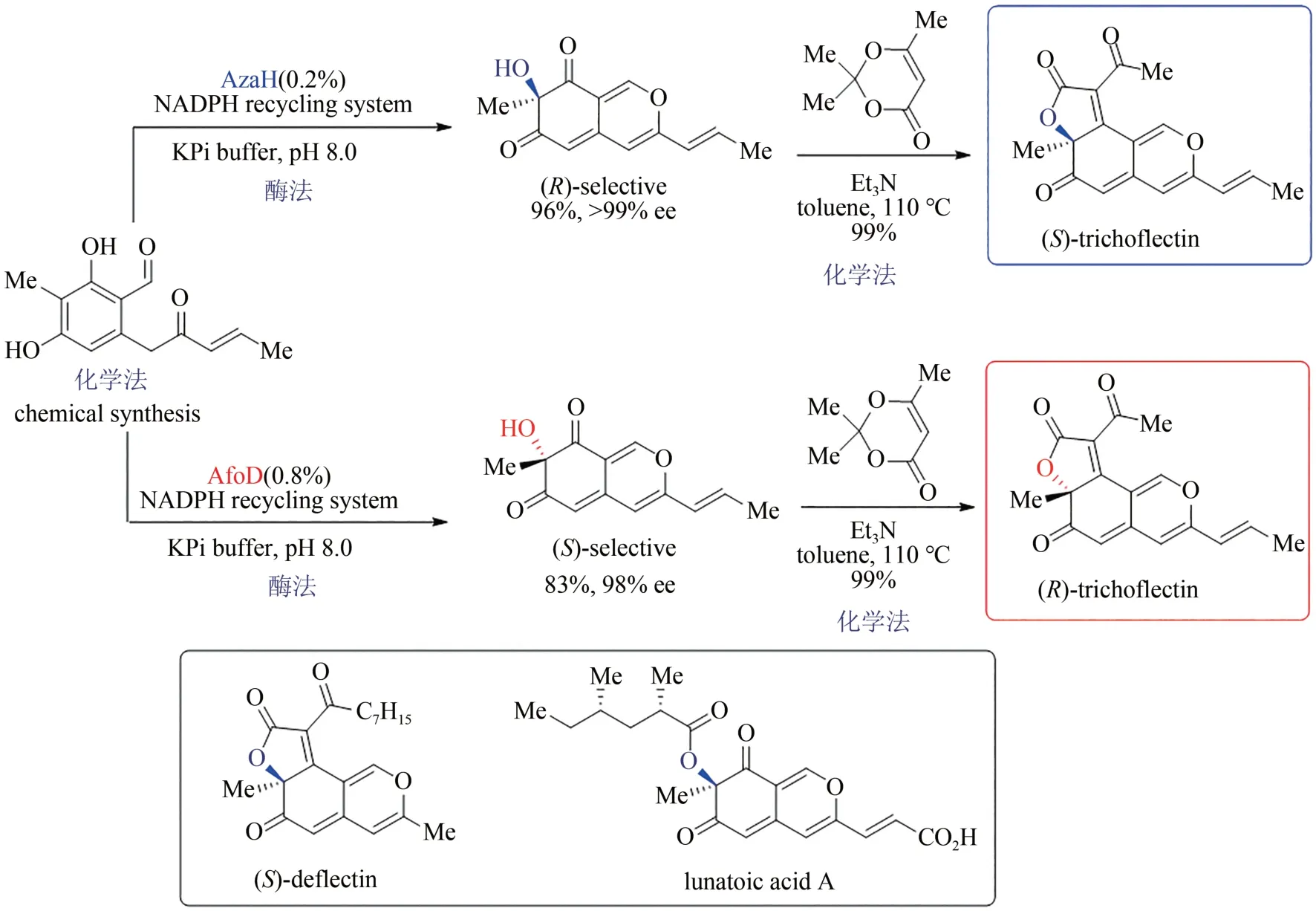
圖13 嗜氮酮天然產物的化學-酶法立體多樣性全合成Fig.13 Stereodivergent and chemoenzymatic synthesis of azaphilone natural products
3.5 卡英酸的化學-酶法全合成
卡英酸(kainic acid)又稱紅藻酸,是一種來源于海洋藻類的天然有機分子,可作為離子型谷氨酸受體抑制劑應用于神經性疾病的治療[108]。盡管該化合物結構并不復雜,但相連的三個手性中心為其立體選擇性合成帶來了很大挑戰,已報道的化學合成路線有70 余條[109],但這些方法的實際應用仍存在巨大困難。2019 年,加州大學Bradley Moore 團隊對產生卡英酸的兩種海藻進行全基因組測序[110],鑒定出兩個關鍵酶:N-異戊二烯基轉移酶KabA 與雙加氧酶KabC,并在大腸桿菌中進行異源表達,嘗試建立卡英酸的生物合成途徑。最后為實現卡英酸的高效制備,作者發展了化學-酶交叉合成體系(圖14):以谷氨酸為起始原料,與3-甲基-2-丁烯醛進行還原胺化反應,以56%收率得到卡英酸前體,再使用純化的DsKabC 酶催化后續的成環步驟,46%收率得到卡英酸;更為重要的是,作者使用大腸桿菌全細胞體系可以57%收率實現卡英酸的克級制備,避免了化學中間體及酶的分離純化,大大提高了合成效率。

圖14 卡英酸的化學-酶法全合成Fig.14 Chemoenzymatic synthesis of kainic acid
3.6 鬼臼毒素的化學-酶法全合成
作為植物來源的木質素類天然代謝產物,鬼臼毒素(podophyllotoxin)及衍生物因其所特有的抗腫瘤、抗病毒等生物活性而備受關注[111]。鬼臼毒素的傳統來源為植物提取,受限于植物生長緩慢,同時過度開采帶來環境破壞等不利因素,不能滿足人類需要。相比之下,近年來利用化學-酶法全合成鬼臼毒素取得了顯著進展。2019 年,Chang Weichen 等報道了一種α-酮戊二酸依賴型雙加氧酶(2-ODD-PH)[112],可催化脫氧鬼臼毒素(deoxypodophyllotoxin)到亞太因(yatein)的碳-碳成鍵反應。基于此發現,Michael Fuchs 和Hans Renata先后發展了兩種化學-酶法合成鬼臼毒素新策略。Michael Fuchs 課題組提供的路線[圖15(a)],首先利用金屬鋅促進胡椒醛與烯丙基溴化物反應制得高烯丙醇中間體,并在銠催化劑作用下合成消旋的羥化亞太因(rac-hydroxyyatein),再用2-ODD-PH 酶催化環化,通過動力學拆分以39%收率及95% ee 對映選擇性得到鬼臼毒素的差向異構體,最后經化學法反轉C7 位點上的羥基手性獲得鬼臼毒素[113]。同時Hans Renata課題組發展了手性底物轉化的合成路線[圖15(b)],第一步采用化學縮合反應制得亞太因,第二步借助2-ODD-PH酶催化亞太因進行環合反應,獲得鬼臼毒素前體,第三步通過化學法在C7 位點進行羥基化反應,從而合成終產物鬼臼毒素,其分離收率達到58%[114]。與化學全合成方法相比,上述兩條化學-生物融合策略在收率及立體控制上均有明顯提升,有望實現鬼臼毒素規模化全合成。
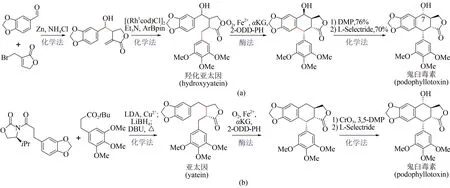
圖15 鬼臼毒素的兩種化學-酶法合成途徑Fig.15 Two chemoenzymatic synthesis pathways of podophyllotoxin
4 結論與展望
藥物開發離不開種類多樣、功能豐富的天然產物。近兩個世紀來,得益于化學分離手段的進步,人類逐漸擺脫了利用動植物/微生物混合提取物的原始用藥方式,并邁向了使用高純度單體天然產物的階段;更為重要的是,隨著有機合成方法和技術的不斷提高,天然產物的化學全合成得到快速全面發展,化學家甚至為眾多藥物活性天然產物分子開發出了多條全合成工藝路線。然而,對很多結構復雜的活性天然產物分子,由于合成步驟多、工藝路線長、選擇性控制難、總收率低等問題,嚴重制約了其實際生產和廣泛應用。隨著分子生物學、系統生物學及合成生物學的快速發展,尤其是定向進化及基因組測序技術的不斷進步,成千上萬的微生物及動植物基因組信息不斷被挖掘,人類已探明生物體內眾多天然產物的全基因合成途徑與酶學機制,這為構建細胞工廠從頭異源合成天然產物提供了便利條件,同時也為合成生物學變革天然產物全合成提供契機。聚酮類、生物堿類、萜類、甾體類、大環內酰胺類、核苷類等復雜天然產物的生物合成已取得較大進展;值得一提的是,我國科學家在牛胰島素、酵母丙氨酸轉移核糖核酸、青蒿素及人參皂苷等多個重要天然產物的生物合成方面也取得多項突破性進展[115],形成了良好的研究基礎。盡管如此,天然產物生物全合成仍存在諸多挑戰性問題亟待解決,如類天然產物新分子的生物合成一直是該領域的重要課題[116-120];含有多個手性中心的立體復雜天然產物的生物合成還存在效率較低、機制不明、途徑優化困難等問題;此外結合金屬催化、光催化、電催化及自由基化學的最新進展,如何實現生物合成與惰性鍵活化及多級串聯反應等新型策略的有機融合,實現天然產物的多樣性全合成也是具有潛力的發展方向。
總之,對天然產物的全合成,無論化學方法還是生物學方法,都各有其特點,如何將兩者優勢互補,相互促進,從而實現更多天然產物高效全合成,是未來重要發展方向;例如廣譜抗癌藥物紫杉醇的微生物異源生物合成,國內外多個團隊已經取得了良好進展,可以獲得三環紫杉二烯(taxadiene)及三環紫杉二烯醇(taxadienols)[121-127],在大腸桿菌中的產量比之前報道提高了萬倍以上,相信不久的將來借助化學和生物互補的途徑定能實現其高效全合成。同時,如何借助當今蓬勃發展的人工智能技術,實現生物元件(如酶元件、啟動子元件等)的挖掘、設計與改造,以及人工生物合成途徑拼裝等過程的智能化、自動化、高效化,助力天然產物異源再造,將成為本領域未來發展的另一新趨勢[128-130]。展望生物合成策略在天然產物及其類似物合成中的應用,更大的進步將會不斷涌現,并為人類健康與社會發展貢獻更大力量。
猜你喜歡
天天愛科學(2022年9期)2022-09-15 01:12:54
天天愛科學(2022年4期)2022-05-23 12:41:48
當代水產(2022年3期)2022-04-26 14:26:56
科學大眾(2021年9期)2021-07-16 07:02:54
軍事文摘(2020年20期)2020-11-28 11:42:50
航空世界(2020年10期)2020-01-19 14:36:20
科技知識動漫(2017年7期)2017-08-09 19:52:45
科技知識動漫(2017年5期)2017-05-11 21:34:16
科技知識動漫(2017年4期)2017-04-15 22:24:55
科技知識動漫(2017年2期)2017-02-06 20:59:46
