固相萃取-液相色譜-串聯質譜法同時測定葛根中11種黃酮苷元的含量
2021-11-29 14:50:34侯建波史穎珠毛壬熠張文華
理化檢驗-化學分冊 2021年10期
侯建波,謝 文,史穎珠,李 杰,汪 鵬,毛壬熠,張文華
(1.杭州海關技術中心,杭州 310016; 2.浙江省檢驗檢疫科學技術研究院,杭州 310016;3.浙江立德產品技術有限公司,杭州 310016; 4.浙江大學 生物系統工程與食品科學學院,杭州 310058)
葛根具有解肌退熱、生津止渴、透疹、升陽止瀉、通經活絡、解酒毒等功效,常用于治療外感發熱頭痛、項背強痛、口渴、消渴、麻疹不透、熱痢、泄瀉、眩暈頭痛、中風偏癱、胸痹心痛和酒毒傷中。2000年葛根被國家衛生部正式批準為藥食兩用植物,可用于新型中藥和功能性保健(食)品中[1]。
以葛根素、大豆苷、染料木素和染料木苷為代表的黃酮類化合物是葛根的主要有效成分,其中含量最高的是葛根素,《中華人民共和國藥典》(2015年版)(簡稱Ch P 2015)將葛根素的含量作為衡量葛根質量的重要指標。目前,葛根中黃酮類化合物含量的測定方法主要有分光光度法[2-4]、液相色譜法[5-11]、液相色譜-串聯質譜法(LC-MS/MS)[12-13]和液相色譜-高分辨質譜法[14-16]等。由于黃酮類化合物種類較多,黃酮苷元可與糖類形成多種黃酮苷而普遍存在于植物體內[17],但各黃酮苷的含量不一,目前除了液相色譜-高分辨質譜法,其他分析手段較難全面獲得黃酮類化合物的信息。
本工作篩選出葛根中11種主要的黃酮苷(葛根素-6″-O-木糖苷、甘草苷、大豆苷、野黃芩苷、大豆黃苷、蘆丁、鳶尾苷、染料木苷、山萘酚-3-O-蕓香糖苷、黃芩苷和刺芒柄花苷)及其對應的黃酮苷元(葛根素、甘草素、大豆素、野黃芩素、大豆黃素、槲皮素、鳶尾黃素、染料木素、山萘酚、黃芩素和刺芒柄花素),通過鹽酸-乙醇溶液的混合液對葛根進行水解,提取其中的黃酮苷元,采用Waters HLB固相萃取柱凈化,建立了LC-MS/MS測定葛根中葛根素、甘草素、大豆素、野黃芩素、大豆黃素、槲皮素、鳶尾黃素、染料木素、山萘酚、黃芩素和刺芒柄花素11種黃酮苷元含量的方法。該方法通過測定水解后樣品中黃酮苷元的含量,可以更全面地反映葛根中各苷元所代表的黃酮類化合物的總量。
1 試驗部分
1.1 儀器與試劑
1100型液相色譜儀;API 4000型三重四極桿串聯質譜儀;Milli-Q Synergy 185型超純水器;IKA MS3 Basic型渦旋器;24孔固相萃取裝置;Heraeus Multifuge X1R型臺式離心機;G&G JJ500型電子天平;Mettler AE260型電子天平。
單標準儲備溶液:10 g·L-1,稱取適量的大豆黃素、大豆素、槲皮素、刺芒柄花素等22種標準品,用甲醇溶解,配制成質量濃度均為10 g·L-1的單標準儲備溶液。
11種黃酮苷元混合標準溶液:100μg·L-1,移取適量的葛根素、甘草素、大豆素、野黃芩素、大豆黃素、槲皮素、鳶尾黃素、染料木素、山萘酚、黃芩素、刺芒柄花素單標準儲備溶液,用甲醇稀釋,配制成質量濃度為100μg·L-1的11種黃酮苷元混合標準溶液。
混合標準溶液:10μg·L-1,移取適量的22種黃酮苷和黃酮苷元單標準儲備溶液,用甲醇稀釋,配制成質量濃度為10μg·L-1的混合標準溶液。
大豆黃素標準品純度為98.2%,大豆素標準品純度為99.0%,槲皮素標準品純度為96.4%,刺芒柄花素標準品純度為98.5%,染料木素標準品純度為99.0%,甘草素標準品純度為96.9%,黃芩素標準品純度為99.2%,野黃芩素標準品純度為96.2%,葛根素標準品純度為87.0%,山萘酚標準品純度為95.7%,鳶尾黃素標準品純度為98.1%,大豆黃苷標準品純度為92.9%,大豆苷標準品純度為93.3%,蘆丁標準品純度為95.0%,刺芒柄花苷標準品純度為99.0%,染料木苷標準品純度為95.9%,甘草苷標準品純度為96.9%,黃芩苷標準品純度為96.8%,野黃芩苷標準品純度為88.9%,葛根素-6″-O-木糖苷標準品純度為100%,山萘酚-3-O-蕓香糖苷標準品純度為99.8%,鳶尾苷標準品純度為98.8%。
甲醇、乙腈為色譜純;甲酸為質譜純;鹽酸為優級純;乙醇、叔丁基對苯二酚(TBHQ)、丁基羥基茴香醚(BHA)、2,6-二叔丁基對甲酚(BHT)、L-(+)-抗壞血酸均為分析純;試驗用水為GB/T 6682規定的一級水。
1.2 儀器工作條件
1)色譜條件 Mightysil RP-18色譜柱(150 mm×4.6 mm,3μm);柱溫 25 ℃;流量0.4 mL·min-1;進樣量20μL;流動相A為甲醇,B為0.15%(體積分數,下同)甲酸溶液,測定大豆苷、大豆黃苷和刺芒柄花苷時B為水。梯度洗脫程序:0~10.0 min時,A 由40%升至75%;10.0~15.0 min時,A 由75%升至90%,保持6.0 min;21.0~23.0 min時,A 由90%下降至40%,保持7.0 min。
2)質譜條件 電噴霧離子(ESI)源,離子源溫度540℃;負離子掃描模式;多反應監測(MRM)模式;電噴霧電壓-4 500 V;霧化氣壓力289 k Pa,氣簾氣壓力310 kPa,輔助氣壓力172 kPa。其他質譜參數見表1,其中,“*”為定量離子。
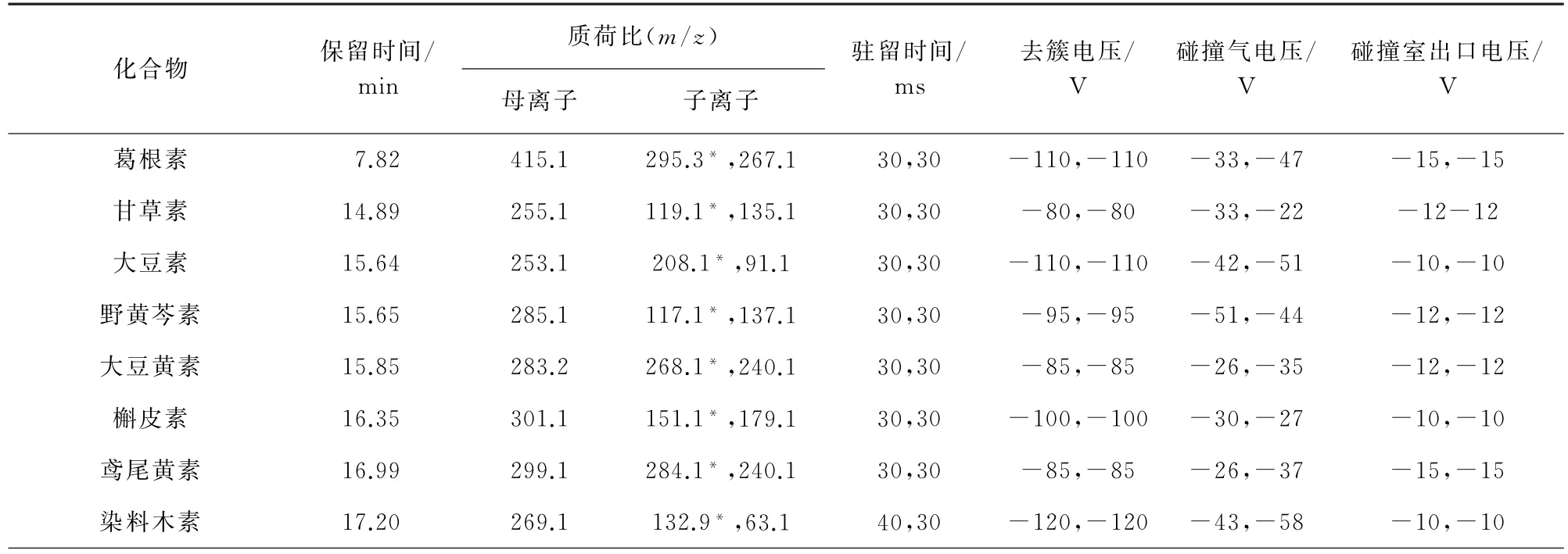
表1 質譜參數Tab.1 MS parameters
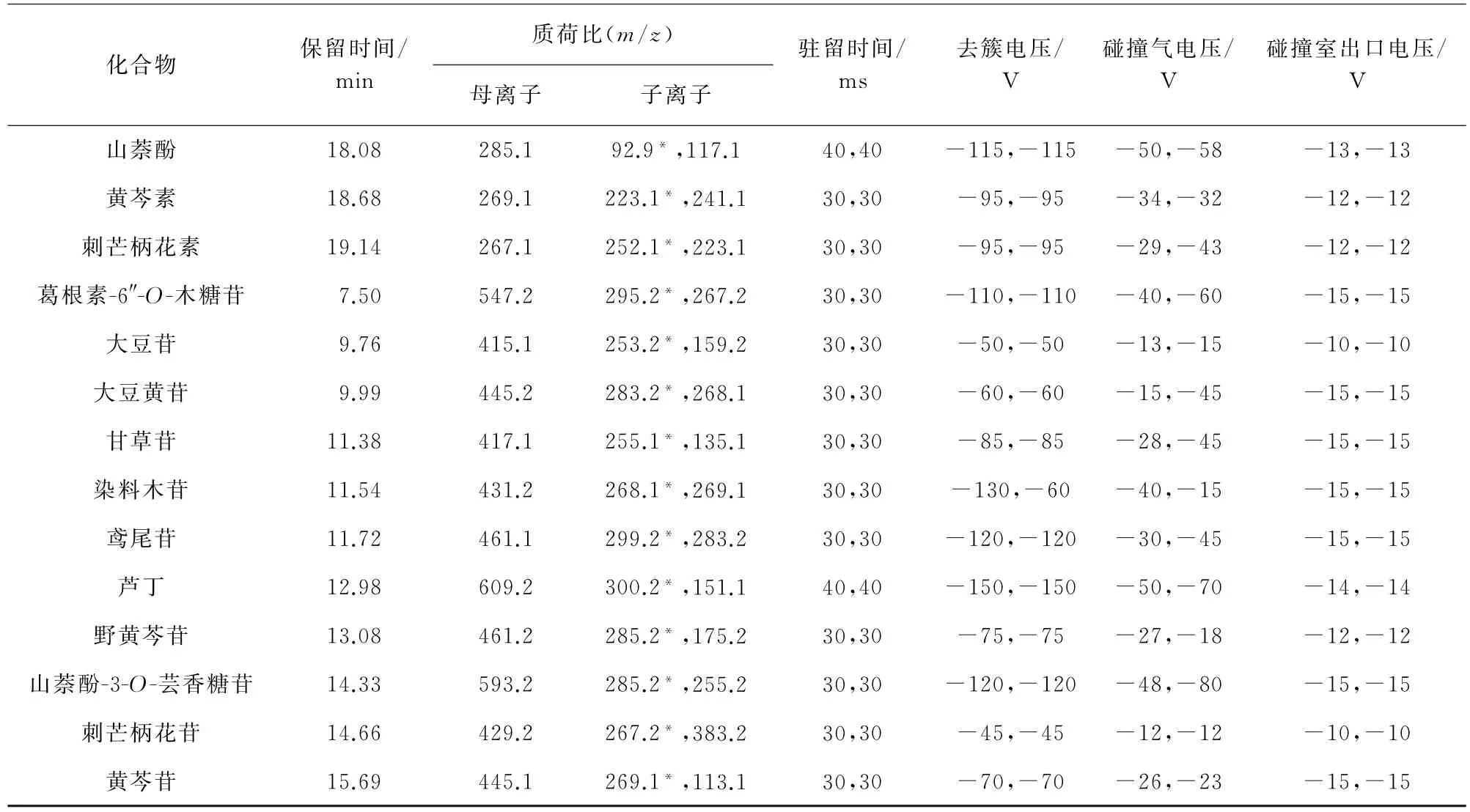
表1(續)
1.3 試驗方法
稱取葛根樣品0.10 g于150 mL圓底燒瓶中,加入0.5 g TBHQ,36 mL 50%(體積分數,下同)乙醇溶液和4 mL鹽酸,在氮氣保護下,水浴加熱回流120 min。水解結束后,冷卻,用水定容至50 mL容量瓶中,取1 mL上清液,加入9 mL水,混合均勻,轉移至經5 mL甲醇和5 mL 5%(體積分數,下同)甲醇溶液活化的 Waters HLB固相萃取柱中,用5 mL 20%(體積分數,下同)甲醇溶液進行淋洗,抽干,再用10 mL甲醇洗脫,控制流量為1~2 mL·min-1,收集全部洗脫液。將洗脫液用甲醇定容至10 mL容量瓶中,搖勻,用0.22μm有機濾膜過濾,取0.1 mL濾液,再用體積比為4∶6的甲醇-0.15%甲酸溶液的混合液定容至10 mL容量瓶中,混合均勻,按儀器工作條件進行測定,外標法定量。
2 結果與討論
2.1 水解條件的選擇
通常采用浸提和回流兩種方式對中草藥中的有效成分進行提取。由于黃酮苷元容易被氧化,參考ChP 2015,試驗考察了TBHQ、BHA、BHT和抗壞血酸等4種不同抗氧化劑對目標物的保護情況。結果顯示:以BHA和BHT為抗氧化劑時,槲皮素、山萘酚、黃岑素的回收率均較低,可能是由于BHA和BHT在乙醇溶液中的溶解性較差,目標物容易被氧化;以抗壞血酸為抗氧化劑時,槲皮素、山萘酚的回收率較低;以TBHQ為抗氧化劑時,槲皮素、山萘酚、黃岑素的回收率均得到明顯提升。進一步試驗發現,當加入0.1 g TBHQ時,槲皮素和山萘酚的回收率均大于70%,而黃芩素的回收率較低;當加入0.5,1.0 g TBHQ 時,黃芩素的回收率可提升至70%左右。考慮到節約成本,試驗選擇抗氧化劑為0.5 g TBHQ。
同時,試驗考察了不同體積分數(30%,50%,70%)的乙醇溶液對黃酮苷水解效果的影響。結果顯示,乙醇體積分數對黃酮苷的水解沒有明顯影響,由于黃酮苷的水溶性較好,黃酮苷元的醇溶性較好,綜合考慮,試驗選擇乙醇體積分數為50%。
在50%乙醇溶液中,加入一定量的鹽酸,使鹽酸體積分數分別為5%,10%,12.5%,并在氮氣保護下水浴加熱回流30,60,90,120,180 min進行試驗。結果顯示,當鹽酸體積分數為10%,水解時間為120 min時,各黃酮苷可完全水解。因此,試驗最終選擇水解條件:抗氧化劑為0.5 g TBHQ,水解溶液為含10%鹽酸的50%乙醇溶液,水解時間為120 min。在上述優化條件下,對10μg·L-1的11種黃酮苷元混合標準溶液進行測定,各黃酮苷元的回收率均大于80%,表明該水解條件下,各黃酮苷元可穩定存在。
2.2 凈化條件的選擇
在LC-MS/MS測定前,通常采用液液萃取、固相萃取、QuEChERS等方式凈化樣品,以降低背景干擾,減少基質效應的影響,從而提高結果的準確性。
按照試驗方法,將水解后的樣品溶液轉移至CNW C18(500 mg/6 mL)、Waters C18(500 mg/6 mL)、Waters HLB(200 mg/6 mL)、Biocomma HLB(200 mg/6 mL)等4種固相萃取柱中,分別采用體積分數為10%,20%,30%,40%,50%的甲醇溶液對其進行淋洗。結果表明:采用C18固相萃取柱凈化,當甲醇體積分數為20%時,僅葛根素被洗脫下來,而其他目標物均不會被洗脫;采用HLB固相萃取柱凈化,當甲醇體積分數為50%時,僅葛根素被洗脫下來,而其他化合物均不會被洗脫。因此,試驗選擇20%甲醇溶液進行淋洗。接著,以甲醇為洗脫液,考察了上述4種固相萃取柱對11種黃酮苷元回收率的影響,結果見圖1。
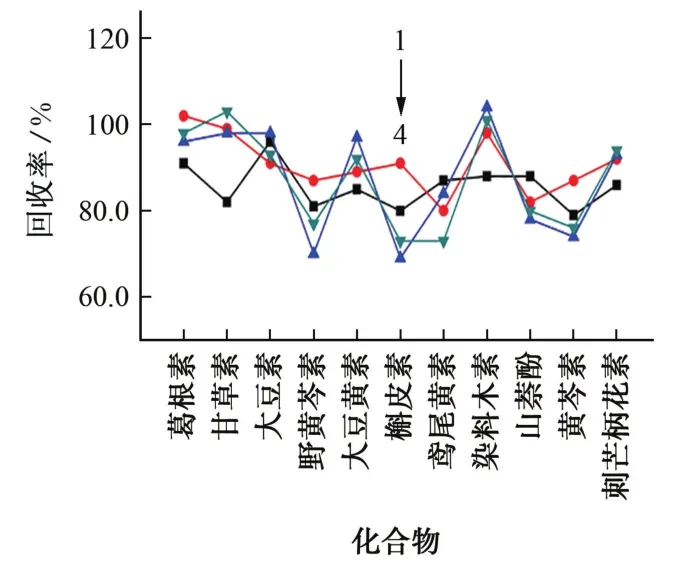
圖1 固相萃取柱對黃酮苷元回收率的影響Fig.1 Effect of solid phase extraction column on recovery of flavonoid aglycones
結果表明,使用HLB固相萃取柱時,各目標物的回收率相對較高,并且Waters HLB固相萃取柱的穩定性較好,因此試驗選擇Waters HLB固相萃取柱,以20%甲醇溶液為淋洗液,以甲醇為洗脫液來進行凈化。
2.3 色譜條件的選擇
按照試驗方法,考察了Mightysil RP-18色譜柱(150 mm×4.6 mm,3μm)和Inertsil ODS-3色譜柱(150 mm×4.6 mm,3μm)對各目標物分離效果的影響。結果表明,以Mightysil RP-18色譜柱為固定相時,各目標物的分離效果相對較好,因此試驗選擇Mightysil RP-18色譜柱。
以Mightysil RP-18色譜柱為固定相,考察了甲醇或乙腈為流動相A,水或0.15%甲酸溶液為流動相B時各目標物的分離情況。結果表明:大部分目標物在甲醇中的色譜峰強度和峰形均優于在乙腈中的,考慮到與前處理凈化的有效銜接,試驗選擇流動相A為甲醇;并且大部分目標物在0.15%甲酸溶液中的色譜峰強度和峰形均優于在水中的,而大豆黃苷以水為流動相B時,其色譜峰強度提高近10倍,大豆苷和刺芒柄花苷以水為流動相B時,其色譜峰強度提高近20倍。因此,對大豆黃苷、大豆苷、刺芒柄花苷進行分析時以水為流動相B,對其他黃酮苷和黃酮苷元進行分析時以0.15%甲酸溶液為流動相B。在上述優化條件下,對10μg·L-1的混合標準溶液進行分析,各目標物的分離情況如圖2所示。
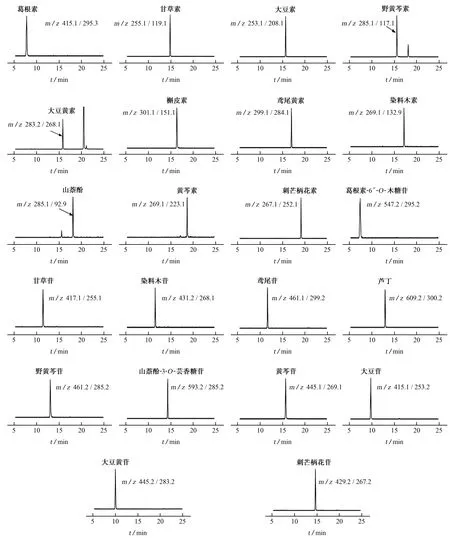
圖2 黃酮苷和黃酮苷元的色譜圖Fig.2 Chromatograms of flavonoid glycosides and flavonoid aglycones
2.4 質譜條件的選擇
試驗對各黃酮苷和黃酮苷元標準溶液進行稀釋,采用流動注射的方式,在負離子模式下進行母離子全掃描,確定準分子離子峰,再以目標物的準分子離子為母離子,對其子離子進行全掃描。按照歐盟EC/657指令的要求,選擇兩個特征子離子,以信噪比高、峰形好、干擾小的離子對作為定量離子對,通過MRM模式優化質譜參數,各黃酮苷和黃酮苷元的質譜參數見表1。
2.5 基質效應
LC-MS/MS測定藥物殘留時,樣品基質對目標物具有增強或抑制效應,從而影響測定結果的準確性。按照試驗方法,對葛根樣品進行處理得到葛根基質溶液,采用體積比為4∶6的甲醇-0.15%甲酸溶液的混合液(溶劑)和葛根基質溶液分別配制10μg·L-1的11種黃酮苷元混合溶液,按照儀器工作條件進樣分析,記錄各目標物在上述兩種溶液中定量離子對的響應強度,并計算離子抑制率[離子抑制率(%)=(基質響應強度-溶劑響應強度)×100%/溶劑響應強度][18],結果見表2。
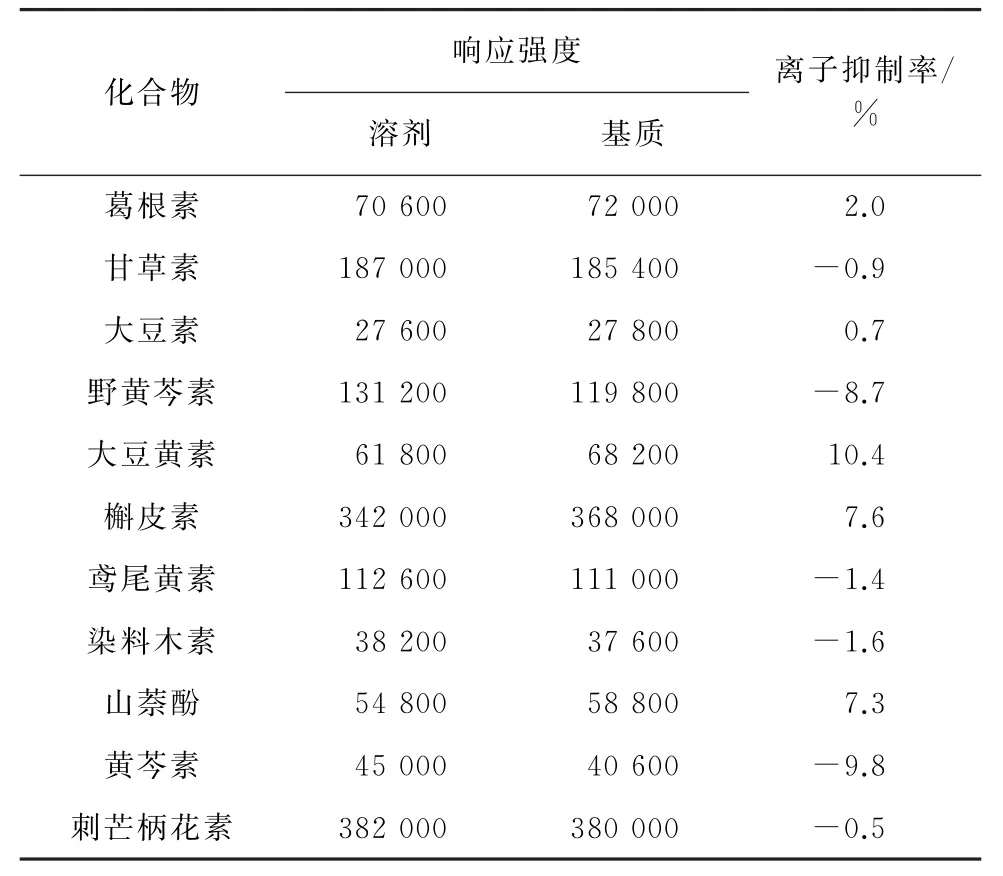
表2 基質效應Tab.2 Matrix effect
結果顯示,各目標物的離子抑制率均小于11%,說明無明顯的基質效應,因此試驗采用體積比為4∶6的甲醇-0.15%甲酸溶液的混合液配制的混合標準溶液系列制作標準曲線。
2.6 標準曲線、檢出限和測定下限
移取適量的11種黃酮苷元混合標準溶液,用體積比為4∶6的甲醇-0.15%甲酸溶液的混合液逐級稀釋,配制成各黃酮苷元質量濃度為1,2,5,10,20,50μg·L-1(相當于黃酮苷元的質量分數為0.05%,0.10%,0.25%,0.50%,1.00%,2.50%)的 混 合 標 準溶液系列,按照儀器工作條件進行測定,外標法定量。以各黃酮苷元的質量分數為橫坐標,其對應的峰面積為縱坐標繪制標準曲線。結果顯示,各黃酮苷元的質量分數在0.05%~2.50%內與其對應的峰面積呈線性關系,線性參數見表3。
以3倍信噪比(S/N)和10倍信噪比計算方法的檢出限(3S/N)和測定下限(10S/N),結果見表3。
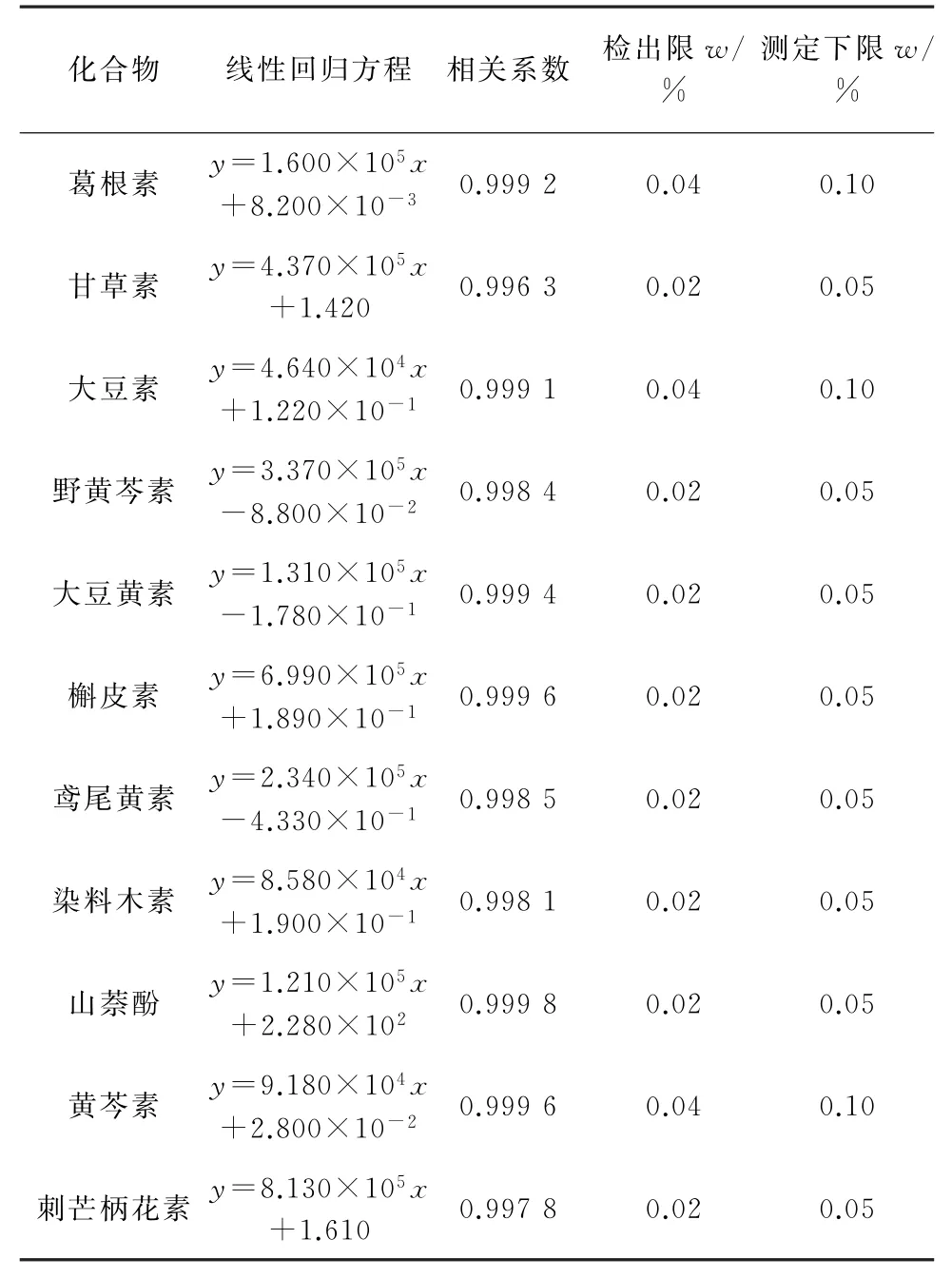
表3 線性參數、檢出限和測定下限Tab.3 Linearity parameters,detection limits and lower limits of determination
2.7 精密度和回收試驗
按照試驗方法對葛根樣品進行3個濃度水平的加標回收試驗(加入各黃酮苷標準溶液,相當于水解后0.25%,0.50%,1.00%的黃酮苷元),每個濃度水平測定6次,計算回收率和測定值的相對標準偏差(RSD),結果見表4。

表4 精密度和回收試驗結果(n=6)Tab.4 Results of tests for precision and recovery(n=6)
由表4可知:黃酮苷元的回收率為75.0%~94.0%,測定值的 RSD為3.5%~12%。
2.8 樣品分析
按照試驗方法分別對經水解和未經水解的10個葛根樣品進行測定,結果見表5。
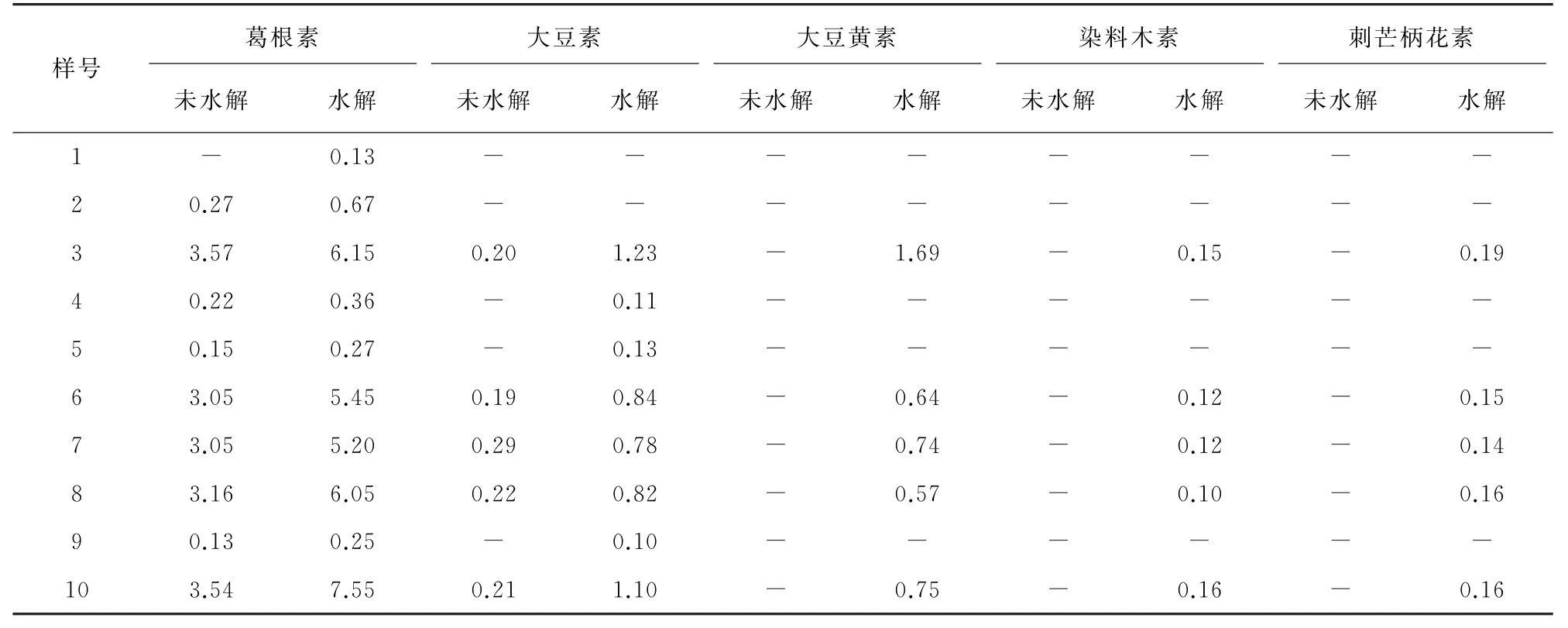
表5 樣品分析結果Tab.5 Analytical results of samples %
結果表明,葛根經水解處理后,甘草素、野黃芩素、槲皮素、鳶尾黃素、山萘酚、黃芩素未被檢出,而葛根素、大豆素、大豆黃素、染料木素和刺芒柄花素的質量分數明顯提高,說明黃酮苷和游離黃酮苷元在葛根中同時存在,本方法通過測定水解后的黃酮苷元,可更全面地反映該苷元所代表黃酮類化合物的含量。
本工作用含10%鹽酸的50%乙醇溶液對葛根中的黃酮苷進行水解得到黃酮苷元,采用 Waters HLB固相萃取柱凈化,建立了LC-MS/MS測定葛根中11種黃酮苷元含量的方法。該方法可更全面地反映葛根中各黃酮苷元所代表的該類黃酮類化合物的含量,對后續黃酮類物質含量及藥物活性關系分析、葛根的品質及品種研究具有重要的輔助作用。
猜你喜歡
今日農業(2022年16期)2022-11-09 23:18:44
中國化肥信息(2022年5期)2022-08-30 01:58:26
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
今日農業(2021年20期)2021-11-26 01:23:56
今日農業(2021年14期)2021-10-14 08:35:34
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
中成藥(2017年8期)2017-11-22 03:19:40
中成藥(2017年10期)2017-11-16 00:50:13
中成藥(2017年4期)2017-05-17 06:09:50
